 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-2022 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
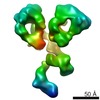
| タイトル | Three-dimensional reconstruction of another targeted individual antibody particle by individual-particle electron tomography (IPET). The reconstruction displayed three ring-shaped domains that corresponding to three domain of IgG antibody. | |||||||||
 マップデータ マップデータ | Single targeted particle structure reconstructed by focus-EM reconstruction algorithm of individual particle electron tomography method | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード |  human IgG antibody (免疫グロブリンG) human IgG antibody (免疫グロブリンG) | |||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||
| 手法 | サブトモグラム平均法 / ネガティブ染色法 / 解像度: 14.6 Å | |||||||||
 データ登録者 データ登録者 | Zhang L / Ren G | |||||||||
 引用 引用 | ジャーナル: PLoS One / 年: 2012 タイトル: IPET and FETR: experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. 著者: Lei Zhang / Gang Ren /  要旨: The dynamic personalities and structural heterogeneity of proteins are essential for proper functioning. Structural determination of dynamic/heterogeneous proteins is limited by conventional ...The dynamic personalities and structural heterogeneity of proteins are essential for proper functioning. Structural determination of dynamic/heterogeneous proteins is limited by conventional approaches of X-ray and electron microscopy (EM) of single-particle reconstruction that require an average from thousands to millions different molecules. Cryo-electron tomography (cryoET) is an approach to determine three-dimensional (3D) reconstruction of a single and unique biological object such as bacteria and cells, by imaging the object from a series of tilting angles. However, cconventional reconstruction methods use large-size whole-micrographs that are limited by reconstruction resolution (lower than 20 Å), especially for small and low-symmetric molecule (<400 kDa). In this study, we demonstrated the adverse effects from image distortion and the measuring tilt-errors (including tilt-axis and tilt-angle errors) both play a major role in limiting the reconstruction resolution. Therefore, we developed a "focused electron tomography reconstruction" (FETR) algorithm to improve the resolution by decreasing the reconstructing image size so that it contains only a single-instance protein. FETR can tolerate certain levels of image-distortion and measuring tilt-errors, and can also precisely determine the translational parameters via an iterative refinement process that contains a series of automatically generated dynamic filters and masks. To describe this method, a set of simulated cryoET images was employed; to validate this approach, the real experimental images from negative-staining and cryoET were used. Since this approach can obtain the structure of a single-instance molecule/particle, we named it individual-particle electron tomography (IPET) as a new robust strategy/approach that does not require a pre-given initial model, class averaging of multiple molecules or an extended ordered lattice, but can tolerate small tilt-errors for high-resolution single "snapshot" molecule structure determination. Thus, FETR/IPET provides a completely new opportunity for a single-molecule structure determination, and could be used to study the dynamic character and equilibrium fluctuation of macromolecules. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_2022.map.gz | 14.3 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-2022-v30.xmlemd-2022.xml | 10.6 KB 10.6 KB | 表示 表示 | EMDBヘッダ |
| 画像 | EMD-2022-v02.tif | 54.8 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2022ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2022 http://ftp.pdbj.org/pub/emdb/structures/EMD-2022ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2022 | HTTPS FTP |
-関連構造データ








-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_2022.map.gz / 形式: CCP4 / 大きさ: 29.8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Single targeted particle structure reconstructed by focus-EM reconstruction algorithm of individual particle electron tomography method | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.406 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Human IgG Antibody
| 全体 | 名称: Human IgG Antibody免疫グロブリンG |
|---|---|
| 要素 |
|
-超分子 #1000: Human IgG Antibody
| 超分子 | 名称: Human IgG Antibody / タイプ: sample / ID: 1000 詳細: The sample was thawed from storage at -80 degrees Celcius before being loaded onto the grid 集合状態: Monomer / Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 160 KDa / 理論値: 160 KDa |
-分子 #1: IgG Antibody
| 分子 | 名称: IgG Antibody / タイプ: protein_or_peptide / ID: 1 / Name.synonym: IgG / コピー数: 1 / 集合状態: Monomer / 組換発現: No / データベース: NCBI |
|---|---|
| 由来(天然) | 生物種: Homo sapiens (ヒト) / 株: IgG / 別称: Human / 細胞中の位置: Plasma |
| 分子量 | 実験値: 160 KDa / 理論値: 160 KDa |
-実験情報
-構造解析
| 手法 | ネガティブ染色法 |
|---|---|
 解析 解析 | サブトモグラム平均法 |
-試料調製
| 濃度 | 0.01 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 詳細: 1X Dulbeccos phosphate-buffered saline (Invitrogen, La Jolla, CA), 2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, and 8.1 mM Na2HPO4 |
| 染色 | タイプ: NEGATIVE 詳細: EM Specimens were prepared by optimized negative-staining EM specimen preparation protocol as described Zhang L. and Ren G, Journal of Lipid Research, (2010) 51, 1228-1236 and (2011) 52, 175- ...詳細: EM Specimens were prepared by optimized negative-staining EM specimen preparation protocol as described Zhang L. and Ren G, Journal of Lipid Research, (2010) 51, 1228-1236 and (2011) 52, 175-84. In brief, antibody was diluted to 0.01 mg/ml with deionized water. Aliquots (about 3ul) were applied to the 200 mesh glow-discharged thin carbon-coated EM grids (Cu-200CN, Pacific Grid-Tech, USA). The grid was washed by deionized water for three times, and then washed by 1% uranyl formate for three times before blotting to drying. |
| グリッド | 詳細: 200 mesh glow-discharged thin carbon-coated EM grids (Cu-200CN, Pacific Grid-Tech, USA) |
| 凍結 | 凍結剤: NONE / 装置: OTHER |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI 20 |
|---|---|
| 電子線 | 加速電圧: 200 kV / 電子線源: LAB6 |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / Cs: 2.0 mm / 最大 デフォーカス(公称値): 2.0 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 80000 |
| 試料ステージ | 試料ホルダー: Gatan / 試料ホルダーモデル: OTHER / Tilt series - Axis1 - Min angle: -60 ° / Tilt series - Axis1 - Max angle: 60 ° |
| 詳細 | tilt step is 1.5 degree |
| 撮影 | カテゴリ: CCD フィルム・検出器のモデル: GATAN ULTRASCAN 4000 (4k x 4k) デジタル化 - サンプリング間隔: 1.406 µm / 実像数: 81 / 平均電子線量: 250 e/Å2 / ビット/ピクセル: 16 |
-画像解析
| CTF補正 | 詳細: TOMOCTF |
|---|---|
| 最終 角度割当 | 詳細: Tomography tilt angle from -60 to 60 in step of 1.5 |
| 最終 再構成 | アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 14.6 Å / 解像度の算出法: OTHER / ソフトウェア - 名称: IPET, and, FETR 詳細: Map was reconstructed by individual-particle electron tomography (IPET)and Focus ET Reconstruction Algorithm. |
| 詳細 | Single targeted particle"s images were reconstructed by focus-EM reconstruction algorithm of individual particle electron tomography method. Average number of tilts used in the 3D reconstructions: 81. Average tomographic tilt angle increment: 1.5. |
