 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xpc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human GGT1 in complex with full GSH molecule | ||||||
 Components Components | (Glutathione hydrolase 1 ... ) x 2 ) x 2 | ||||||
 Keywords Keywords | HYDROLASE / Substrate-ENZYME COMPLEX / NTN-HYDROLASE FAMILY / GLYCOPROTEIN / N-GLYCOSYLATION / CELL SURFACE | ||||||
| Function / homology |  Function and homology informationleukotriene-C4 hydrolase / peptidyltransferase activity / leukotriene-C(4) hydrolase / leukotriene D4 biosynthetic process / Defective GGT1 causes GLUTH / Defective GGT1 in aflatoxin detoxification causes GLUTH / LTC4-CYSLTR mediated IL4 production / Glutathione synthesis and recycling / peptide modification / gamma-glutamyltransferase ...leukotriene-C4 hydrolase / peptidyltransferase activity / leukotriene-C(4) hydrolase / leukotriene D4 biosynthetic process / Defective GGT1 causes GLUTH / Defective GGT1 in aflatoxin detoxification causes GLUTH / LTC4-CYSLTR mediated IL4 production / Glutathione synthesis and recycling / peptide modification / gamma-glutamyltransferase / glutathione gamma-glutamate hydrolase / glutathione hydrolase activity / leukotriene C4 gamma-glutamyl transferase activity / glutathione catabolic process / glutamate metabolic process / glutathione biosynthetic process / leukotriene metabolic process / cysteine biosynthetic process / Aflatoxin activation and detoxification / Synthesis of Leukotrienes (LT) and Eoxins (EX) / regulation of immune system process / Paracetamol ADME / zymogen activation / amino acid metabolic process / fatty acid metabolic process / regulation of inflammatory response / spermatogenesis / proteolysis / extracellular space / extracellular exosome / plasma membrane Function and homology informationleukotriene-C4 hydrolase / peptidyltransferase activity / leukotriene-C(4) hydrolase / leukotriene D4 biosynthetic process / Defective GGT1 causes GLUTH / Defective GGT1 in aflatoxin detoxification causes GLUTH / LTC4-CYSLTR mediated IL4 production / Glutathione synthesis and recycling / peptide modification / gamma-glutamyltransferase ...leukotriene-C4 hydrolase / peptidyltransferase activity / leukotriene-C(4) hydrolase / leukotriene D4 biosynthetic process / Defective GGT1 causes GLUTH / Defective GGT1 in aflatoxin detoxification causes GLUTH / LTC4-CYSLTR mediated IL4 production / Glutathione synthesis and recycling / peptide modification / gamma-glutamyltransferase / glutathione gamma-glutamate hydrolase / glutathione hydrolase activity / leukotriene C4 gamma-glutamyl transferase activity / glutathione catabolic process / glutamate metabolic process / glutathione biosynthetic process / leukotriene metabolic process / cysteine biosynthetic process / Aflatoxin activation and detoxification / Synthesis of Leukotrienes (LT) and Eoxins (EX) / regulation of immune system process / Paracetamol ADME / zymogen activation / amino acid metabolic process / fatty acid metabolic process / regulation of inflammatory response / spermatogenesis / proteolysis / extracellular space / extracellular exosome / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.26 Å | ||||||
 Authors Authors | Terzyan, S.S. / Hanigan, M. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2020 Title: Crystal structures of glutathione- and inhibitor-bound human GGT1: critical interactions within the cysteinylglycine binding site. Authors: Terzyan, S.S. / Nguyen, L.T. / Burgett, A.W.G. / Heroux, A. / Smith, C.A. / You, Y. / Hanigan, M.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xpc.cif.gz | 128.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xpc.ent.gz | 95.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6xpc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xp/6xpcftp://data.pdbj.org/pub/pdb/validation_reports/xp/6xpc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6xpbC  4gdxS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Glutathione hydrolase 1 ... , 2 types, 2 molecules AB
| #1: Protein | / GGT 1 / Gamma-glutamyltransferase 1 / Glutathione hydrolase 1 / Leukotriene-C4 hydrolase Mass: 38533.664 Da / Num. of mol.: 1 / Mutation: V272A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GGT1, GGT / Plasmid: PPICZAA / Production host:  Komagataella pastoris (fungus) / Strain (production host): X-33 Komagataella pastoris (fungus) / Strain (production host): X-33References: UniProt: P19440, glutathione gamma-glutamate hydrolase, gamma-glutamyltransferase, leukotriene-C4 hydrolase |
|---|---|
| #2: Protein | / GGT 1 / Gamma-glutamyltransferase 1 / Glutathione hydrolase 1 / Leukotriene-C4 hydrolase Mass: 20014.438 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GGT1, GGT / Plasmid: PPICZAA / Production host: Komagataella pastoris (fungus) / Strain (production host): X-33References: UniProt: P19440, glutathione gamma-glutamate hydrolase, gamma-glutamyltransferase, leukotriene-C4 hydrolase |
-Sugars , 1 types, 6 molecules 
| #3: Sugar | ChemComp-NAG / N-Acetylglucosamine Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 6 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 6Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
|---|
-Non-polymers , 4 types, 251 molecules 

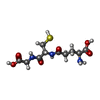

| #4: Chemical | Chloride Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#5: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#6: Chemical | ChemComp-GSH / | Glutathione Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S / Feature type: SUBJECT OF INVESTIGATION Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S / Feature type: SUBJECT OF INVESTIGATION#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 247 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.93 Å3/Da / Density % sol: 58.06 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 20-25%PEG3350, 0.1M ammonium cloride, 0.1M sodium cacodilate pH 6.0 Temp details: Room temperature |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL / Beamline: BL12-2 / Wavelength: 0.9792 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 17, 2016 / Details: mirror |
| Radiation | Monochromator: SI 111 DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 2.26→20 Å / Num. obs: 32583 / % possible obs: 98.8 % / Observed criterion σ(I): -3 / Redundancy: 6.47 % / Biso Wilson estimate: 47.9 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 15.01 |
| Reflection shell | Resolution: 2.26→2.39 Å / Redundancy: 6.08 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 2.52 / Num. unique obs: 4931 / % possible all: 93.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 4GDX Resolution: 2.26→19.96 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.951 / SU B: 6.361 / SU ML: 0.147 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.216 / ESU R Free: 0.184 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 129.05 Å2 / Biso mean: 42.886 Å2 / Biso min: 13.94 Å2
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.26→19.96 Å
| |||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.26→2.318 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
