 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5u89: Crystal structure of a cross-module fragment from the dimodular N... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5u89 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a cross-module fragment from the dimodular NRPS DhbF | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/INHIBITOR /  nonribosomal peptide synthetase / MbtH-like protein / mechanism-based inhibitor / megaenzyme / HYDROLASE-INHIBITOR complex nonribosomal peptide synthetase / MbtH-like protein / mechanism-based inhibitor / megaenzyme / HYDROLASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationbiosynthetic process / hydrolase activity, acting on ester bonds / phosphopantetheine bindingSimilarity search - Function | ||||||
| Biological species |  Geobacillus sp. (bacteria) Geobacillus sp. (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.075 Å | ||||||
 Authors Authors | Tarry, M.J. / Schmeing, T.M. | ||||||
| Funding support |  Canada, 1items Canada, 1items
| ||||||
 Citation Citation | Journal: Structure / Year: 2017 Title: X-Ray Crystallography and Electron Microscopy of Cross- and Multi-Module Nonribosomal Peptide Synthetase Proteins Reveal a Flexible Architecture. Authors: Michael J Tarry / Asfarul S Haque / Khanh Huy Bui / T Martin Schmeing / Abstract: Nonribosomal peptide synthetases (NRPS) are macromolecular machines that produce peptides with diverse activities. Structural information exists for domains, didomains, and even modules, but little ...Nonribosomal peptide synthetases (NRPS) are macromolecular machines that produce peptides with diverse activities. Structural information exists for domains, didomains, and even modules, but little is known about higher-order organization. We performed a multi-technique study on constructs from the dimodular NRPS DhbF. We determined a crystal structure of a cross-module construct including the adenylation (A) and peptidyl carrier protein (PCP) domains from module 1 and the condensation domain from module 2, complexed with an adenosine-vinylsulfonamide inhibitor and an MbtH-like protein (MLP). The action of the inhibitor and the role of the MLP were investigated using adenylation reactions and isothermal titration calorimetry. In the structure, the PCP and A domains adopt a novel conformation, and noncovalent, cross-module interactions are limited. We calculated envelopes of dimodular DhbF using negative-stain electron microscopy. The data show large conformational variability between modules. Together, our results suggest that NRPSs lack a uniform, rigid supermodular architecture. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5u89.cif.gz | 407.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5u89.ent.gz | 337.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5u89.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/u8/5u89ftp://data.pdbj.org/pub/pdb/validation_reports/u8/5u89 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 121345.070 Da / Num. of mol.: 1 / Fragment: UNP residues 437-1504 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus sp. (bacteria) / Strain: Y4.1MC1 / Gene: GY4MC1_0171 / Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: A0A0F6BHX2, UniProt: A0A7U3YCA6*PLUS |
|---|---|
| #2: Protein | Mass: 9246.290 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus sp. (bacteria) / Strain: Y4.1MC1 / Gene: GY4MC1_0172 / Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: A0A0F6BHX3, UniProt: A0A7U3YBY3*PLUS |
| #3: Chemical | ChemComp-MJ8 / 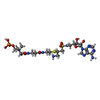  Mass: 743.747 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H42N9O12PS2 Mass: 743.747 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H42N9O12PS2 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.65 Å3/Da / Density % sol: 73.57 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 5.5 Details: 1.6 M ammonium sulfate, 0.1 M Bis-Tris pH 5, 2 mM EDTA, 0.008% (v/v) octyl-beta-D galactoside |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI / Beamline: 08ID-1 / Wavelength: 0.9795 Å |
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Oct 7, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 3.075→58.56 Å / Num. obs: 47171 / % possible obs: 98.56 % / Redundancy: 2 % / Rmerge(I) obs: 0.06869 / Rsym value: 0.06869 / Net I/σ(I): 5.04 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3.075→58.564 Å / SU ML: 0.44 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 26.03 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.075→58.564 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
