 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5tfp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the SETDB2 Amino Terminal Domain | ||||||
 Components Components | Histone-lysine N-methyltransferase SETDB2 | ||||||
 Keywords Keywords |  TRANSFERASE / lysine methyltransferase / helical handshake motif / dimerization domain TRANSFERASE / lysine methyltransferase / helical handshake motif / dimerization domain | ||||||
| Function / homology |  Function and homology information: / positive regulation of DNA methylation-dependent heterochromatin formation / left/right axis specification / histone H3K9 methyltransferase activity / heart looping / heterochromatin organization / Transferases; Transferring one-carbon groups; Methyltransferases / chromosome segregation / PKMTs methylate histone lysines / chromosome ...: / positive regulation of DNA methylation-dependent heterochromatin formation / left/right axis specification / histone H3K9 methyltransferase activity / heart looping / heterochromatin organization / Transferases; Transferring one-carbon groups; Methyltransferases / chromosome segregation / PKMTs methylate histone lysines / chromosome / mitotic cell cycle / cell division / negative regulation of gene expression / negative regulation of DNA-templated transcription / DNA binding / zinc ion binding / nucleoplasm / nucleus / cytosol Function and homology information: / positive regulation of DNA methylation-dependent heterochromatin formation / left/right axis specification / histone H3K9 methyltransferase activity / heart looping / heterochromatin organization / Transferases; Transferring one-carbon groups; Methyltransferases / chromosome segregation / PKMTs methylate histone lysines / chromosome ...: / positive regulation of DNA methylation-dependent heterochromatin formation / left/right axis specification / histone H3K9 methyltransferase activity / heart looping / heterochromatin organization / Transferases; Transferring one-carbon groups; Methyltransferases / chromosome segregation / PKMTs methylate histone lysines / chromosome / mitotic cell cycle / cell division / negative regulation of gene expression / negative regulation of DNA-templated transcription / DNA binding / zinc ion binding / nucleoplasm / nucleus / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2 Å | ||||||
 Authors Authors | Khorasanizadeh, S. / Kim, Y. | ||||||
 Citation Citation | Journal: To Be Published Title: Identification of a helical dimerization domain in SETDB2 lysine methyltransferase Authors: Khorasanizadeh, S. / Kim, Y. / Osborne, T.F. / Rastinejad, F. / Potluri, N. / Su, X. / Roqueta-Rivera, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5tfp.cif.gz | 66.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5tfp.ent.gz | 54.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5tfp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tf/5tfpftp://data.pdbj.org/pub/pdb/validation_reports/tf/5tfp | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




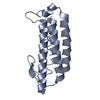
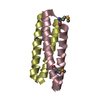




-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 7413.893 Da / Num. of mol.: 2 / Fragment: UNP residues 1-64 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SETDB2, C13orf4, CLLD8, KMT1F / Production host:  Escherichia coli (E. coli) Escherichia coli (E. coli)References: UniProt: Q96T68, histone-lysine N-methyltransferase#2: Chemical | ChemComp-GOL / Glycerol  Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical | ChemComp-TLA / | Tartaric acid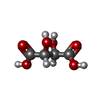  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.08 Å3/Da / Density % sol: 60.07 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 / Details: 0.2 M ammonium tartrate (pH 6.0) and 20% PEG3350 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97931 Å / Beamline: 19-ID / Wavelength: 0.97931 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Feb 13, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97931 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Limit h max: 30 / Limit h min: 0 / Limit k max: 17 / Limit k min: 0 / Limit l max: 60 / Limit l min: 0 / Number: 12742 / D res high: 1.97 Å / D res low: 60.166 Å / Num. obs: 12061 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 12089 / % possible obs: 98.8 % / Redundancy: 8 % / Rmerge(I) obs: 0.08 / Net I/av σ(I): 22.741 / Net I/σ(I): 8.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection scale | Group code: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: SAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2→34.74 Å / Cross valid method: FREE R-VALUE
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 104.98 Å2 / Biso mean: 31.1359 Å2 / Biso min: 9.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→34.74 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.09 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
