 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5lx9: CRYSTAL STRUCTURE OF HUMAN ADIPONECTIN RECEPTOR 2 IN COMPLEX WITH... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5lx9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN ADIPONECTIN RECEPTOR 2 IN COMPLEX WITH A C18 FREE FATTY ACID AT 2.4 ANGSTROM RESOLUTION | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  MEMBRANE PROTEIN / PROGESTIN AND ADIPOQ RECEPTOR FAMILY / INTEGRAL MEMBRANE PROTEIN / 7TM / CERAMIDASE MEMBRANE PROTEIN / PROGESTIN AND ADIPOQ RECEPTOR FAMILY / INTEGRAL MEMBRANE PROTEIN / 7TM / CERAMIDASE | ||||||
| Function / homology |  Function and homology informationadiponectin binding / adipokinetic hormone receptor activity / adiponectin-activated signaling pathway / AMPK inhibits chREBP transcriptional activation activity / vascular wound healing / fatty acid oxidation / hormone-mediated signaling pathway / glucose homeostasis / signaling receptor activity / positive regulation of cold-induced thermogenesis ...adiponectin binding / adipokinetic hormone receptor activity / adiponectin-activated signaling pathway / AMPK inhibits chREBP transcriptional activation activity / vascular wound healing / fatty acid oxidation / hormone-mediated signaling pathway / glucose homeostasis / signaling receptor activity / positive regulation of cold-induced thermogenesis / identical protein binding / metal ion binding / plasma membrane Function and homology informationadiponectin binding / adipokinetic hormone receptor activity / adiponectin-activated signaling pathway / AMPK inhibits chREBP transcriptional activation activity / vascular wound healing / fatty acid oxidation / hormone-mediated signaling pathway / glucose homeostasis / signaling receptor activity / positive regulation of cold-induced thermogenesis ...adiponectin binding / adipokinetic hormone receptor activity / adiponectin-activated signaling pathway / AMPK inhibits chREBP transcriptional activation activity / vascular wound healing / fatty acid oxidation / hormone-mediated signaling pathway / glucose homeostasis / signaling receptor activity / positive regulation of cold-induced thermogenesis / identical protein binding / metal ion binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Mus musculus (house mouse) Mus musculus (house mouse) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Vasiliauskaite-Brooks, I. / Leyrat, C. / Hoh, F. / Granier, S. | ||||||
| Funding support |  France, 1items France, 1items
| ||||||
 Citation Citation | Journal: Nature / Year: 2017 Title: Structural insights into adiponectin receptors suggest ceramidase activity. Authors: Vasiliauskaite-Brooks, I. / Sounier, R. / Rochaix, P. / Bellot, G. / Fortier, M. / Hoh, F. / De Colibus, L. / Bechara, C. / Saied, E.M. / Arenz, C. / Leyrat, C. / Granier, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5lx9.cif.gz | 239.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5lx9.ent.gz | 198.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5lx9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lx/5lx9ftp://data.pdbj.org/pub/pdb/validation_reports/lx/5lx9 | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein / Antibody , 2 types, 2 molecules AH
| #1: Protein | Mass: 35020.805 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: Q86V24*PLUS Spodoptera frugiperda (fall armyworm) / References: UniProt: Q86V24*PLUS |
|---|---|
| #2: Antibody | Mass: 29841.535 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mus musculus (house mouse) / Cell line (production host): Schneider 2 / Production host: Drosophila melanogaster (fruit fly) |
-Non-polymers , 4 types, 541 molecules 
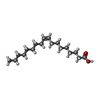

| #3: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||
|---|---|---|---|
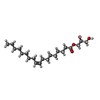
| #4: Chemical | ChemComp-OLA / Oleic acid Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 | ||
| #5: Chemical | ChemComp-OLB / (  Mass: 356.540 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C21H40O4 Mass: 356.540 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C21H40O4#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 527 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.68 Å3/Da / Density % sol: 66.58 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase Details: 30-45% PEG 400, 0.1 M HEPES pH 7.0, and 50-100 mM potassium citrate, 0.01 mM AdipoRon |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF / Beamline: ID30B / Wavelength: 0.976251 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Jun 30, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976251 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→101.13 Å / Num. obs: 33584 / % possible obs: 100 % / Redundancy: 10.5 % / Biso Wilson estimate: 41.55 Å2 / CC1/2: 0.988 / Rmerge(I) obs: 0.527 / Net I/σ(I): 5.5 |
| Reflection shell | Resolution: 2.4→2.5 Å / Redundancy: 10.2 % / Rmerge(I) obs: 5.681 / Mean I/σ(I) obs: 1.1 / CC1/2: 0.489 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.4→101.13 Å / Cor.coef. Fo:Fc: 0.878 / Cor.coef. Fo:Fc free: 0.894 / SU R Cruickshank DPI: 0.256 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.294 / SU Rfree Blow DPI: 0.195 / SU Rfree Cruickshank DPI: 0.187
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 50.14 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.32 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→101.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.47 Å / Rfactor Rfree error: 0 / Total num. of bins used: 17
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
