 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4tvj | ||||||
|---|---|---|---|---|---|---|---|
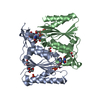
| Title | HUMAN ARTD2 (PARP2) - CATALYTIC DOMAIN IN COMPLEX WITH OLAPARIB | ||||||
 Components Components | Poly [ADP-ribose] polymerase 2 | ||||||
 Keywords Keywords |  TRANSFERASE / Poly(ADP-Ribose) transferase / Inhibitor / ADP-Ribosylation TRANSFERASE / Poly(ADP-Ribose) transferase / Inhibitor / ADP-Ribosylation | ||||||
| Function / homology |  Function and homology information Function and homology informationhippocampal neuron apoptotic process / response to oxygen-glucose deprivation / poly-ADP-D-ribose binding / positive regulation of cell growth involved in cardiac muscle cell development / NAD+-protein-serine ADP-ribosyltransferase activity / NAD DNA ADP-ribosyltransferase activity / NAD+- protein-aspartate ADP-ribosyltransferase activity / NAD+-protein-glutamate ADP-ribosyltransferase activity / DNA ADP-ribosylation / HDR through MMEJ (alt-NHEJ) ...hippocampal neuron apoptotic process / response to oxygen-glucose deprivation / poly-ADP-D-ribose binding / positive regulation of cell growth involved in cardiac muscle cell development / NAD+-protein-serine ADP-ribosyltransferase activity / NAD DNA ADP-ribosyltransferase activity / NAD+- protein-aspartate ADP-ribosyltransferase activity / NAD+-protein-glutamate ADP-ribosyltransferase activity / DNA ADP-ribosylation / HDR through MMEJ (alt-NHEJ) / poly-ADP-D-ribose modification-dependent protein binding / DNA repair-dependent chromatin remodeling / NAD+ ADP-ribosyltransferase / protein auto-ADP-ribosylation / protein poly-ADP-ribosylation / site of DNA damage / NAD+-protein ADP-ribosyltransferase activity / decidualization / Transferases; Glycosyltransferases; Pentosyltransferases / NAD+ ADP-ribosyltransferase activity / POLB-Dependent Long Patch Base Excision Repair / nucleosome binding / extrinsic apoptotic signaling pathway / nucleotidyltransferase activity / DNA Damage Recognition in GG-NER / base-excision repair / Dual Incision in GG-NER / Formation of Incision Complex in GG-NER / double-strand break repair / damaged DNA binding / DNA repair / DNA damage response / chromatin binding / nucleolus / nucleoplasm / nucleusSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Karlberg, T. / Thorsell, A.G. / Ekblad, T. / Pinto, A.F. / Schuler, H. | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2017 Title: Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. Authors: Thorsell, A.G. / Ekblad, T. / Karlberg, T. / Low, M. / Pinto, A.F. / Tresaugues, L. / Moche, M. / Cohen, M.S. / Schuler, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4tvj.cif.gz | 291.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4tvj.ent.gz | 236.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4tvj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tv/4tvjftp://data.pdbj.org/pub/pdb/validation_reports/tv/4tvj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4r5wC  4r6eC  4rv6C  4undC  4uxbC  5lx6C  3kczS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41827.922 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PARP2, ADPRT2, ADPRTL2 / Plasmid: PNIC28-BSA4 / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): R3 PRARE / References: UniProt: Q9UGN5, NAD+ ADP-ribosyltransferase Escherichia coli BL21(DE3) (bacteria) / Strain (production host): R3 PRARE / References: UniProt: Q9UGN5, NAD+ ADP-ribosyltransferase#2: Chemical | Olaparib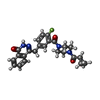  Mass: 434.463 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H23FN4O3 / Comment: medication, inhibitor*YM Mass: 434.463 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H23FN4O3 / Comment: medication, inhibitor*YM#3: Chemical | Glycerol  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 267 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 267 / Source method: isolated from a natural source / Formula: H2OSequence details | The sequence mismatch is due to sequence conflicts between Uniprot and GenBank (ID AF085734.1). In ...The sequence mismatch is due to sequence conflicts between Uniprot and GenBank (ID AF085734.1). In this case authors have used the sequence from GenBank (http://www.ncbi.nlm.nih.gov/nuccore/4808556), here residue 447 is Histidine. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.74 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8.5 / Details: 25% PEG3350, 0.1M Tris |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9919 Å / Beamline: I04 / Wavelength: 0.9919 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jul 27, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9919 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. all: 45851 / Num. obs: 45851 / % possible obs: 99.5 % / Redundancy: 7.5 % / Rmerge(I) obs: 0.109 / Rsym value: 0.107 / Net I/σ(I): 14.6 |
| Reflection shell | Resolution: 2.1→2.15 Å / Redundancy: 7.3 % / Rmerge(I) obs: 0.669 / Mean I/σ(I) obs: 3.1 / % possible all: 98.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3KCZ Resolution: 2.1→29.28 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.924 / SU B: 10.101 / SU ML: 0.127 / Cross valid method: THROUGHOUT / ESU R: 0.22 / ESU R Free: 0.183 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.879 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.1→29.28 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|