 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qm8: Crystal Structure of a Putative Cysteine Dioxygnase From Bacillus... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qm8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of a Putative Cysteine Dioxygnase From Bacillus subtilis: A Alternative Modeling of 3EQE | ||||||
 Components Components | Cysteine dioxygenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Cupin Fold / Cysteine dioxygnease | ||||||
| Function / homology |  Function and homology informationcysteine dioxygenase / cysteine dioxygenase activity / L-cysteine catabolic process / oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen / ferrous iron binding Function and homology informationcysteine dioxygenase / cysteine dioxygenase activity / L-cysteine catabolic process / oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen / ferrous iron bindingSimilarity search - Function | ||||||
| Biological species |  Bacillus subtilis subsp. subtilis (bacteria) Bacillus subtilis subsp. subtilis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 2.82 Å | ||||||
 Authors Authors | Hartman, S.H. / Driggers, C.M. / Karplus, P.A. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2015 Title: Structures of Arg- and Gln-type bacterial cysteine dioxygenase homologs. Authors: Driggers, C.M. / Hartman, S.J. / Karplus, P.A. | ||||||
| History |
| ||||||
| Remark 0 | THIS ENTRY 4QM8 REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA R3EQESF DETERMINED ...THIS ENTRY 4QM8 REFLECTS AN ALTERNATIVE MODELING OF THE ORIGINAL STRUCTURAL DATA R3EQESF DETERMINED BY AUTHORS OF THE PDB ENTRY 3EQE:S.M.VOROBIEV,M.SU,J.SEETHARAMAN,J.BENACH,F.FOROUHAR, G.CLAYTON,B.COOPER,H.WANG,E.L.FOOTE,C.CICCOSANTI,L.MAO, R.XIAO,T.B.ACTON,G.T.MONTELIONE,J.F.HUNT,L.TONG,NORTHEAST STRUCTURAL GENOMICS CONSORTIUM (NESG) |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qm8.cif.gz | 134.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qm8.ent.gz | 111.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4qm8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qm/4qm8ftp://data.pdbj.org/pub/pdb/validation_reports/qm/4qm8 | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 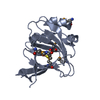
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / CDO Mass: 19895.480 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus subtilis subsp. subtilis (bacteria)Strain: 168 / Gene: BSU31140, cdoA, Cysteine Dioxygenase, yubC / Plasmid: pET21 / Production host: Escherichia coli (E. coli) / Strain (production host): Bl21(de3) / References: UniProt: O32085, cysteine dioxygenase#2: Chemical | Iron  Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe#3: Chemical | Cysteine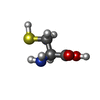  Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7NO2S Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7NO2S#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | AUTHORS HAVE DECLARED THAT THE ACTIVE SITE DENSITY AT THIS RESOLUTION IS NOT DEFINITIVE, SO TO BE ...AUTHORS HAVE DECLARED THAT THE ACTIVE SITE DENSITY AT THIS RESOLUTION | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54.18 % / Description: AUTHOR USED THE SF DATA FROM ENTRY 3EQE. |
|---|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.82→46.51 Å / SU ML: 0.39 / σ(F): 1.98 / Phase error: 26.94 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.82→46.51 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
