 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4agu | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE HUMAN CDKL1 KINASE DOMAIN | ||||||
 Components Components | CYCLIN-DEPENDENT KINASE-LIKE 1 | ||||||
 Keywords Keywords |  TRANSFERASE / PHOSPHO-MIMETIC / KINASE TRANSFERASE / PHOSPHO-MIMETIC / KINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationciliary transition zone / regulation of cilium assembly / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / heart development / protein phosphorylation / intracellular membrane-bounded organelle / extracellular exosome / nucleoplasm / ATP binding ...ciliary transition zone / regulation of cilium assembly / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / heart development / protein phosphorylation / intracellular membrane-bounded organelle / extracellular exosome / nucleoplasm / ATP binding / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Canning, P. / Sharpe, T.D. / Allerston, C. / Savitsky, P. / Pike, A.C.W. / Muniz, J.R.C. / Chaikuad, A. / Kuo, K. / Burgess-Brown, N. / Ayinampudi, V. ...Canning, P. / Sharpe, T.D. / Allerston, C. / Savitsky, P. / Pike, A.C.W. / Muniz, J.R.C. / Chaikuad, A. / Kuo, K. / Burgess-Brown, N. / Ayinampudi, V. / Zhang, Y. / Thangaratnarajah, C. / Ugochukwu, E. / Vollmar, M. / Krojer, T. / Weigelt, J. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / von Delft, F. / Knapp, S. / Bullock, A. | ||||||
 Citation Citation | Journal: Cell Rep / Year: 2018 Title: CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Authors: Canning, P. / Park, K. / Goncalves, J. / Li, C. / Howard, C.J. / Sharpe, T.D. / Holt, L.J. / Pelletier, L. / Bullock, A.N. / Leroux, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4agu.cif.gz | 351 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4agu.ent.gz | 284.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4agu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ag/4aguftp://data.pdbj.org/pub/pdb/validation_reports/ag/4agu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3zduC  4aaaSC  4bbmC  4bgqC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 36306.891 Da / Num. of mol.: 3 / Fragment: KINASE DOMAIN, RESIDUES 1-300 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Description: MAMMALIAN GENE COLLECTION / Plasmid: PNIC-CTHF / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 (DE3) / Variant (production host): R3 PRARE2 / References: UniProt: Q00532 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 (DE3) / Variant (production host): R3 PRARE2 / References: UniProt: Q00532#2: Chemical | 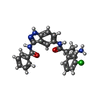  Mass: 447.917 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C24H22ClN5O2 Mass: 447.917 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C24H22ClN5O2#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 315 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 315 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, THR 159 TO ASP ENGINEERED RESIDUE IN CHAIN A, TYR 161 TO GLU ...ENGINEERED | Sequence details | CONFLICT Q274E IS A DOCUMENTED NATURAL VARIANT. CONFLICT A152 AND N301D ARE DOCUMENTED SEQUENCE ...CONFLICT Q274E IS A DOCUMENTED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 41 % / Description: NONE |
|---|---|
| Crystal grow | Details: 17.5% PEG 3350, 0.1 M (NH4)CL PH 6.3, 0.05 M MG FORMATE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I24 / Wavelength: 0.9778 / Beamline: I24 / Wavelength: 0.9778 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Aug 5, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9778 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→29.78 Å / Num. obs: 33165 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 4.8 % / Biso Wilson estimate: 54.57 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 8.1 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 4.7 % / Rmerge(I) obs: 0.69 / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4AAA Resolution: 2.4→29.78 Å / Cor.coef. Fo:Fc: 0.9582 / Cor.coef. Fo:Fc free: 0.936 / SU R Cruickshank DPI: 0.479 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.446 / SU Rfree Blow DPI: 0.239 / SU Rfree Cruickshank DPI: 0.245 Details: RESIDUES 152-163 OF CHAIN A AND RESIDUE 39-40 OF CHAIN B ARE DISORDERED. IDEAL-DIST CONTACT TERM SETUP. ALL ATOMS HAVE CCP4 ATOM TYPE FROM LIBRARY. NCS REPRESENTATION IS RESTRAINT LSSR (- ...Details: RESIDUES 152-163 OF CHAIN A AND RESIDUE 39-40 OF CHAIN B ARE DISORDERED. IDEAL-DIST CONTACT TERM SETUP. ALL ATOMS HAVE CCP4 ATOM TYPE FROM LIBRARY. NCS REPRESENTATION IS RESTRAINT LSSR (-AUTONCS). NUMBER OF RESTRAIN LIBRARIES USED 9.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 60.66 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.305 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→29.78 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.47 Å / Total num. of bins used: 17
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
