- PDB-3vnn: Crystal Structure of a sub-domain of the nucleotidyltransferase (... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 3vnn
Title
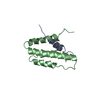
Crystal Structure of a sub-domain of the nucleotidyltransferase (adenylation) domain of human DNA ligase IV
Components
DNA ligase 4
Keywords
LIGASE / DNA ligase / non-homologous end joining / DNA repair / XRCC4
Function / homology
Function and homology information
DNA ligation involved in DNA recombination / positive regulation of chromosome organization / DNA ligase IV complex / DNA ligation involved in DNA repair / DNA ligase activity / DN2 thymocyte differentiation / DNA ligase (ATP) / T cell receptor V(D)J recombination / pro-B cell differentiation / DNA ligase (ATP) activity ...DNA ligation involved in DNA recombination / positive regulation of chromosome organization / DNA ligase IV complex / DNA ligation involved in DNA repair / DNA ligase activity / DN2 thymocyte differentiation / DNA ligase (ATP) / T cell receptor V(D)J recombination / pro-B cell differentiation / DNA ligase (ATP) activity / DNA-dependent protein kinase-DNA ligase 4 complex / single strand break repair / immunoglobulin V(D)J recombination / nonhomologous end joining complex / DNA ligation / V(D)J recombination / double-strand break repair via classical nonhomologous end joining / isotype switching / nucleotide-excision repair, DNA gap filling / positive regulation of neurogenesis / DNA biosynthetic process / cellular response to lithium ion / 2-LTR circle formation / somatic stem cell population maintenance / ligase activity / response to X-ray / chromosome organization / condensed chromosome / neurogenesis / central nervous system development / stem cell proliferation / cellular response to ionizing radiation / response to gamma radiation / Nonhomologous End-Joining (NHEJ) / double-strand break repair via nonhomologous end joining / establishment of integrated proviral latency / double-strand break repair / positive regulation of fibroblast proliferation / T cell differentiation in thymus / fibroblast proliferation / neuron apoptotic process / in utero embryonic development / negative regulation of neuron apoptotic process / cell population proliferation / chromosome, telomeric region / cell cycle / cell division / intracellular membrane-bounded organelle / magnesium ion binding / DNA binding / nucleoplasm / ATP binding / nucleus Similarity search - Function
DNA ligase IV domain / DNA ligase IV / DNA ligase 4 / DNA Ligase 4, adenylation domain / DNA ligase, ATP-dependent / DNA ligase, ATP-dependent, N-terminal / DNA ligase, ATP-dependent, N-terminal domain superfamily / DNA ligase N terminus / DNA ligase/mRNA capping enzyme / ATP-dependent DNA ligase signature 2. ...DNA ligase IV domain / DNA ligase IV / DNA ligase 4 / DNA Ligase 4, adenylation domain / DNA ligase, ATP-dependent / DNA ligase, ATP-dependent, N-terminal / DNA ligase, ATP-dependent, N-terminal domain superfamily / DNA ligase N terminus / DNA ligase/mRNA capping enzyme / ATP-dependent DNA ligase signature 2. / ATP-dependent DNA ligase AMP-binding site. / DNA ligase, ATP-dependent, C-terminal / ATP dependent DNA ligase C terminal region / DNA ligase, ATP-dependent, conserved site / ATP-dependent DNA ligase family profile. / DNA ligase, ATP-dependent, central / ATP dependent DNA ligase domain / BRCA1 C Terminus (BRCT) domain / D-amino Acid Aminotransferase; Chain A, domain 1 / breast cancer carboxy-terminal domain / BRCT domain profile. / BRCT domain / BRCT domain superfamily / Nucleic acid-binding, OB-fold / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Resolution: 2.9→2.97 Å / Redundancy: 11.7 % / Rmerge(I) obs: 0.544 / Mean I/σ(I) obs: 5.19 / Rsym value: 0.544 / % possible all: 100
-
Processing
Software
Name
Version
Classification
MxCuBE
datacollection
SOLVE
phasing
PHENIX
(phenix.refine: 1.7.3_928)
refinement
DENZO
datareduction
SCALEPACK
datascaling
Refinement
Method to determine structure: SAD / Resolution: 2.903→30.64 Å / FOM work R set: 0.7276 / SU ML: 0.29 / σ(F): 1.33 / Phase error: 30.64 / Stereochemistry target values: ML
Rfactor
Num. reflection
% reflection
Rfree
0.3049
381
10 %
Rwork
0.2733
-
-
obs
0.2767
3811
98.88 %
all
-
2263
-
Solvent computation
Shrinkage radii: 0.73 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 95.262 Å2 / ksol: 0.318 e/Å3
Displacement parameters
Biso mean: 109.8132 Å2
Baniso -1
Baniso -2
Baniso -3
1-
8.8971 Å2
0 Å2
-0 Å2
2-
-
8.8971 Å2
0 Å2
3-
-
-
-17.7942 Å2
Refinement step
Cycle: LAST / Resolution: 2.903→30.64 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
949
0
0
3
952
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.014
970
X-RAY DIFFRACTION
f_angle_d
1.971
1319
X-RAY DIFFRACTION
f_dihedral_angle_d
18.975
331
X-RAY DIFFRACTION
f_chiral_restr
0.156
147
X-RAY DIFFRACTION
f_plane_restr
0.011
173
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.9028-3.3224
0.3909
121
0.3042
1100
X-RAY DIFFRACTION
100
3.3224-4.1842
0.2978
126
0.2581
1127
X-RAY DIFFRACTION
100
4.1842-30.6413
0.292
134
0.2742
1203
X-RAY DIFFRACTION
98
Refinement TLS params.
Method: refined / Origin x: -0.1544 Å / Origin y: 12.152 Å / Origin z: 62.2802 Å
11
12
13
21
22
23
31
32
33
T
1.1422 Å2
-0.5319 Å2
0.4325 Å2
-
0.6554 Å2
-0.1712 Å2
-
-
0.6698 Å2
L
3.3707 °2
-0.9543 °2
1.4246 °2
-
1.3123 °2
0.7508 °2
-
-
2.7432 °2
S
0.3848 Å °
0.0965 Å °
0.1399 Å °
-0.0758 Å °
0.1461 Å °
0.004 Å °
-1.1068 Å °
0.6403 Å °
-0.2819 Å °
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
Auth asym-ID
Auth seq-ID
1
X-RAY DIFFRACTION
1
all
A
268 - 406
2
X-RAY DIFFRACTION
1
all
A
501 - 503
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components
 Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj





 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.9765 Å
/ Beamline: ID14-1 / Wavelength: 0.9765 Å Processing
Processing