 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3q4u: Crystal structure of the ACVR1 kinase domain in complex with LDN-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3q4u | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the ACVR1 kinase domain in complex with LDN-193189 | ||||||
 Components Components | Activin receptor type-1 | ||||||
 Keywords Keywords |  TRANSFERASE / Structural Genomics Consortium / SGC / Protein kinase TRANSFERASE / Structural Genomics Consortium / SGC / Protein kinase | ||||||
| Function / homology |  Function and homology information Function and homology informationendocardial cushion cell fate commitment / mitral valve morphogenesis / endocardial cushion fusion / BMP receptor complex / atrial septum primum morphogenesis / BMP receptor activity / cardiac muscle cell fate commitment / acute inflammatory response / activin receptor activity, type I / positive regulation of cardiac epithelial to mesenchymal transition ...endocardial cushion cell fate commitment / mitral valve morphogenesis / endocardial cushion fusion / BMP receptor complex / atrial septum primum morphogenesis / BMP receptor activity / cardiac muscle cell fate commitment / acute inflammatory response / activin receptor activity, type I / positive regulation of cardiac epithelial to mesenchymal transition / positive regulation of determination of dorsal identity / transforming growth factor beta receptor activity, type I / activin receptor complex / endocardial cushion formation / smooth muscle cell differentiation / receptor protein serine/threonine kinase / transmembrane receptor protein serine/threonine kinase activity / pharyngeal system development / activin binding / cellular response to BMP stimulus / activin receptor signaling pathway / negative regulation of activin receptor signaling pathway / transforming growth factor beta binding / embryonic heart tube morphogenesis / gastrulation with mouth forming second / dorsal/ventral pattern formation / determination of left/right symmetry / neural crest cell migration / atrioventricular valve morphogenesis / branching involved in blood vessel morphogenesis / negative regulation of G1/S transition of mitotic cell cycle / ventricular septum morphogenesis / SMAD binding / germ cell development / positive regulation of SMAD protein signal transduction / peptide hormone binding / mesoderm formation / regulation of ossification / BMP signaling pathway / positive regulation of bone mineralization / positive regulation of osteoblast differentiation / negative regulation of signal transduction / transforming growth factor beta receptor signaling pathway / protein tyrosine kinase binding / negative regulation of extrinsic apoptotic signaling pathway / cellular response to growth factor stimulus / positive regulation of peptidyl-tyrosine phosphorylation / apical part of cell / heart development / in utero embryonic development / protein kinase activity / positive regulation of cell migration / cadherin binding / phosphorylation / protein serine/threonine kinase activity / positive regulation of DNA-templated transcription / protein homodimerization activity / positive regulation of transcription by RNA polymerase II / ATP binding / metal ion binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.82 Å | ||||||
 Authors Authors | Chaikuad, A. / Sanvitale, C. / Cooper, C.D.O. / Mahajan, P. / Daga, N. / Petrie, K. / Alfano, I. / Gileadi, O. / Fedorov, O. / Allerston, C.K. ...Chaikuad, A. / Sanvitale, C. / Cooper, C.D.O. / Mahajan, P. / Daga, N. / Petrie, K. / Alfano, I. / Gileadi, O. / Fedorov, O. / Allerston, C.K. / Krojer, T. / Vollmar, M. / von Delft, F. / Weigelt, J. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Plos One / Year: 2013 Title: A new class of small molecule inhibitor of BMP signaling. Authors: Sanvitale, C.E. / Kerr, G. / Chaikuad, A. / Ramel, M.C. / Mohedas, A.H. / Reichert, S. / Wang, Y. / Triffitt, J.T. / Cuny, G.D. / Yu, P.B. / Hill, C.S. / Bullock, A.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3q4u.cif.gz | 516.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3q4u.ent.gz | 424.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3q4u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q4/3q4uftp://data.pdbj.org/pub/pdb/validation_reports/q4/3q4u | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 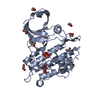 3mtfC  3h9rS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34537.633 Da / Num. of mol.: 4 / Fragment: kinase domain, UNP residues 201-499 / Mutation: Q207D Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ACVR1, ACVRLK2 / Plasmid: pFB-LIC-Bse / Cell line (production host): SF9 / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: Q04771, receptor protein serine/threonine kinase#2: Chemical | ChemComp-LDN / LDN193189  Mass: 406.482 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C25H22N6 Mass: 406.482 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C25H22N6#3: Chemical | ChemComp-EDO / Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-FLC / Citric acid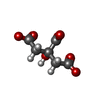  Mass: 189.100 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C6H5O7#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 1019 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1019 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.66 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, sitting drop / pH: 5 Details: 20% PEG 3350, 0.2M ammonium citrate dibasic pH 5.0, VAPOR DIFFUSION, SITTING DROP, temperature 293.15K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 Å / Beamline: I03 / Wavelength: 0.9763 Å |
| Detector | Type: ADSC Q315 3x3 CCD / Detector: CCD / Date: Jan 20, 2010 / Details: Kirkpatrick Baez bimorph mirror pair |
| Radiation | Monochromator: Si (111) double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 1.82→41.88 Å / Num. all: 107198 / Num. obs: 107172 / % possible obs: 99.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.3 % / Biso Wilson estimate: 24.1 Å2 / Rmerge(I) obs: 0.086 / Net I/σ(I): 11.6 |
| Reflection shell | Resolution: 1.82→1.92 Å / Redundancy: 4.2 % / Rmerge(I) obs: 0.734 / Mean I/σ(I) obs: 2 / Num. unique all: 15312 / % possible all: 97.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb id: 3h9r chain A Resolution: 1.82→37.22 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.936 / SU B: 5.525 / SU ML: 0.087 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 2 / ESU R Free: 0.134 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN USED IN REFINEMENT BUT NOT OUTPUT TO PDB
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.583 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.19 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.82→37.22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.82→1.867 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
