 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3m6w: Multi-site-specific 16S rRNA methyltransferase RsmF from Thermus ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3m6w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Multi-site-specific 16S rRNA methyltransferase RsmF from Thermus thermophilus in space group P21212 in complex with S-Adenosyl-L-Methionine | ||||||
 Components Components | rRNA methylase | ||||||
 Keywords Keywords |  TRANSFERASE / rRNA methyltransferase / 5-methylcytidine / RsmF / AdoMet / multi-specific / methyltransferase TRANSFERASE / rRNA methyltransferase / 5-methylcytidine / RsmF / AdoMet / multi-specific / methyltransferase | ||||||
| Function / homology |  Function and homology information16S rRNA (cytosine1407-C5)-methyltransferase / RNA methyltransferase activity / RNA methylation / Transferases; Transferring one-carbon groups; Methyltransferases / rRNA processing / RNA binding / cytoplasm Function and homology information16S rRNA (cytosine1407-C5)-methyltransferase / RNA methyltransferase activity / RNA methylation / Transferases; Transferring one-carbon groups; Methyltransferases / rRNA processing / RNA binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.3 Å | ||||||
 Authors Authors | Demirci, H. / Larsen, H.G.L. / Hansen, T. / Rasmussen, A. / Cadambi, A. / Gregory, S.T. / Kirpekar, F. / Jogl, G. | ||||||
 Citation Citation | Journal: Rna / Year: 2010 Title: Multi-site-specific 16S rRNA methyltransferase RsmF from Thermus thermophilus. Authors: Demirci, H. / Larsen, L.H. / Hansen, T. / Rasmussen, A. / Cadambi, A. / Gregory, S.T. / Kirpekar, F. / Jogl, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3m6w.cif.gz | 210.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3m6w.ent.gz | 163.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3m6w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m6/3m6wftp://data.pdbj.org/pub/pdb/validation_reports/m6/3m6w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3m6uSC  3m6vC  3m6xC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 50922.590 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: RsmF minus / Source: (gene. exp.) Thermus thermophilus (bacteria) / Strain: HB8 / Gene: TTHA1387 / Plasmid: pLJ102 / Production host: Escherichia coli (E. coli) / Strain (production host): CP79References: UniProt: Q5SII2, Transferases; Transferring one-carbon groups; Methyltransferases |
|---|---|
| #2: Chemical | ChemComp-SAM / S-Adenosyl methionine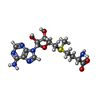  Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S |
| #3: Chemical | ChemComp-CL / Chloride  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 856 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 856 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.74 % |
|---|---|
| Crystal grow | Temperature: 277 K / pH: 8.5 Details: 10% w/v PEG1000, 200 mM NaCl and 100 mM TRIS-HCl (pH8.5), microbatch under oil, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Wavelength: 0.98 Å / Beamline: X4A / Wavelength: 0.98 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Dec 3, 2008 Details: Slits: Variable vertical and fixed horizontal slits. Mirror: Mirror system consisting of two vertically stacked, fused silica, spherical mirrors, to provide vertical focusing and harmonic ...Details: Slits: Variable vertical and fixed horizontal slits. Mirror: Mirror system consisting of two vertically stacked, fused silica, spherical mirrors, to provide vertical focusing and harmonic rejection. One of the mirrors is rhodium coated and the other is uncoated. Located ~19.7 m from source. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit beam condition. Located ~18 m from source ...Monochromator: KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit beam condition. Located ~18 m from source and ~6 m from sample position. Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 5.2 % / Av σ(I) over netI: 24.3 / Number: 633624 / Rmerge(I) obs: 0.058 / Χ2: 1.02 / D res high: 1.29 Å / D res low: 30 Å / Num. obs: 121660 / % possible obs: 95.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.29→30 Å / Num. obs: 121660 / % possible obs: 95.6 % / Redundancy: 5.2 % / Rmerge(I) obs: 0.058 / Χ2: 1.019 / Net I/σ(I): 12.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3M6U Resolution: 1.3→29.597 Å / Occupancy max: 1 / Occupancy min: 0.23 / FOM work R set: 0.893 / SU ML: 0.17 / σ(F): 1.33 / Phase error: 17.56 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 39.211 Å2 / ksol: 0.347 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 84.79 Å2 / Biso mean: 19.302 Å2 / Biso min: 7.47 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→29.597 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 30
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
