 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
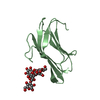
Yorodumi- PDB-3cjs: Minimal Recognition Complex between PrmA and Ribosomal Protein L11 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3cjs | ||||||
|---|---|---|---|---|---|---|---|
| Title | Minimal Recognition Complex between PrmA and Ribosomal Protein L11 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/RIBOSOMAL PROTEIN / S-Adenosyl-L-Methionine dependent methyltransferase /  post-translational modification / multi-specific trimethylation / Ribonucleoprotein / Ribosomal protein / RNA-binding / rRNA-binding / TRANSFERASE-RIBOSOMAL PROTEIN COMPLEX post-translational modification / multi-specific trimethylation / Ribonucleoprotein / Ribosomal protein / RNA-binding / rRNA-binding / TRANSFERASE-RIBOSOMAL PROTEIN COMPLEX | ||||||
| Function / homology |  Function and homology informationhistone methyltransferase activity / Transferases; Transferring one-carbon groups; Methyltransferases / large ribosomal subunit rRNA binding / methylation / ribosome / structural constituent of ribosome / ribonucleoprotein complex / translation / cytosol Function and homology informationhistone methyltransferase activity / Transferases; Transferring one-carbon groups; Methyltransferases / large ribosomal subunit rRNA binding / methylation / ribosome / structural constituent of ribosome / ribonucleoprotein complex / translation / cytosolSimilarity search - Function | ||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.37 Å | ||||||
 Authors Authors | Demirci, H. / Gregory, S.T. / Dahlberg, A.E. / Jogl, G. | ||||||
 Citation Citation | Journal: Structure / Year: 2008 Title: Multiple-Site Trimethylation of Ribosomal Protein L11 by the PrmA Methyltransferase. Authors: Demirci, H. / Gregory, S.T. / Dahlberg, A.E. / Jogl, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3cjs.cif.gz | 57.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3cjs.ent.gz | 42.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3cjs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cj/3cjsftp://data.pdbj.org/pub/pdb/validation_reports/cj/3cjs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3cjqC  3cjrC  3cjtC  3egvC  2nxcS  2nxnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Ribosome / L11 Mtase Mass: 6785.558 Da / Num. of mol.: 1 / Fragment: N-Terminal Domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermus thermophilus (bacteria) / Strain: HB8 / Gene: prmA / Plasmid: pET26b / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 (DE3) StarReferences: UniProt: Q84BQ9, Transferases; Transferring one-carbon groups; Methyltransferases | ||||
|---|---|---|---|---|---|
| #2: Protein | Mass: 7516.837 Da / Num. of mol.: 2 / Fragment: N-Terminal Domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermus thermophilus (bacteria) / Strain: HB8 / Gene: rplK, rpl11 / Plasmid: pET11a / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 (DE3) prmA::TC / References: UniProt: P36238#3: Chemical | Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.22 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 22% w/v PEG3350, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Wavelength: 0.9797 Å / Beamline: X4A / Wavelength: 0.9797 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 15, 2006 Details: Variable vertical and fixed horizontal slits. KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit ...Details: Variable vertical and fixed horizontal slits. KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit beam condition. Located ~18 m from source and ~6 m from sample position. Mirror system consisting of two vertically stacked, fused silica, spherical mirrors, to provide vertical focusing and harmonic rejection. One of the mirrors is rhodium coated and the other is uncoated. Located ~19.7 m from source. |
| Radiation | Monochromator: KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit beam condition. Located ~18 m from source ...Monochromator: KOHZU double crystal monochromator with a water-cooled flat first crystal and a sagittally focused second crystal positioned for a fixed exit beam condition. Located ~18 m from source and ~6 m from sample position. Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9797 Å / Relative weight: 1 |
| Reflection | Resolution: 1.37→30 Å / Num. obs: 37200 / % possible obs: 95.1 % / Observed criterion σ(I): -3 / Redundancy: 3.2 % / Rmerge(I) obs: 0.053 / Net I/σ(I): 22.5 |
| Reflection shell | Resolution: 1.37→1.42 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.386 / Mean I/σ(I) obs: 2.6 / Num. unique all: 3269 / % possible all: 84.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entries 2NXC, 2NXN Resolution: 1.37→25.72 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.951 / SU B: 1.874 / SU ML: 0.039 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.063 / ESU R Free: 0.065 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.711 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.37→25.72 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.37→1.403 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
