 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2y6z: Crystallographic structure of GM23 an example of Catalytic migrat... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2y6z | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystallographic structure of GM23 an example of Catalytic migration from TIM to thiamin phosphate synthase. | ||||||
 Components Components | TRIOSE-PHOSPHATE ISOMERASE Triosephosphate isomerase Triosephosphate isomerase | ||||||
 Keywords Keywords | ISOMERASE | ||||||
| Function / homology |  Function and homology informationglycosome / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytosol / cytoplasm Function and homology informationglycosome / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  TRYPANOSOMA BRUCEI BRUCEI (eukaryote) TRYPANOSOMA BRUCEI BRUCEI (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Saab-Rincon, G. / Olvera, L. / Olvera, M. / Rudino-Pinera, E. / Soberon, X. / Morett, E. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2012 Title: Evolutionary Walk between (Beta/Alpha)(8) Barrels: Catalytic Migration from Triosephosphate Isomerase to Thiamin Phosphate Synthase. Authors: Saab-Rincon, G. / Olvera, L. / Olvera, M. / Rudino-Pinera, E. / Benites, E. / Soberon, X. / Morett, E. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2y6z.cif.gz | 118.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2y6z.ent.gz | 92.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2y6z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y6/2y6zftp://data.pdbj.org/pub/pdb/validation_reports/y6/2y6z | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2y70C  1triS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













































-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Triosephosphate isomerase / GM23 / TIM Mass: 27158.004 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Details: THIS PROTEIN IS RESULT OF ARTIFICIAL MUTATIONS IN ORDER TO OBTAIN A PROTEIN WITH THIAMIN PHOSPHATE SYNTHASE ACTIVITY STARTING FROM A TIM ACTIVITY. Source: (gene. exp.) TRYPANOSOMA BRUCEI BRUCEI (eukaryote)Description: THE GENE FOR THE ENGINEERED GM23 WERE ORIGINALLY OBTAINED FROM TRYPANOSOMA BRUCEI Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): CM1061 THIE- / References: UniProt: P04789, triose-phosphate isomerase ESCHERICHIA COLI (E. coli) / Strain (production host): CM1061 THIE- / References: UniProt: P04789, triose-phosphate isomerase |
|---|---|
| #2: Chemical | ChemComp-TPS / 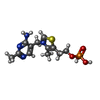  Mass: 345.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H18N4O4PS Mass: 345.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H18N4O4PS |
| #3: Chemical | ChemComp-POP / Pyrophosphate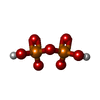  Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | ENGINEERED RESIDUE IN CHAIN A, SER 2 TO GLY ENGINEERED RESIDUE IN CHAIN A, SER 17 TO GLY ENGINEERED ...ENGINEERED |
| Sequence details | THE SEQUENCE DEPOSITED IN THIS ENTRY IS 88.1 PER CENT IDENTICAL TO UNIPROT P04789 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.29 Å3/Da / Density % sol: 71.3 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN WAS CRYSTALLIZED FROM 2 M LI2SO4, 100 MM MES, PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X6A / Wavelength: 0.9795 / Beamline: X6A / Wavelength: 0.9795 |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jan 25, 2006 Details: DOUBLE CRYSTAL CHANNEL CUT, SI(111), 1M LONG RH COATED TOROIDAL MIRROR FOR VERTICAL AND HORIZONTAL FOCUSING |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→34 Å / Num. obs: 15823 / % possible obs: 99 % / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Biso Wilson estimate: 50.84 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 19.6 |
| Reflection shell | Resolution: 2.6→2.76 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.5 / Mean I/σ(I) obs: 2.8 / % possible all: 98.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TRI Resolution: 2.6→33.992 Å / SU ML: 0.43 / σ(F): 1.34 / Phase error: 25.4 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 71.848 Å2 / ksol: 0.37 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 57.8 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→33.992 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
