 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dkw: CRYSTAL STRUCTURE OF TRIOSE-PHOSPHATE ISOMERASE WITH MODIFIED SUB... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dkw | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TRIOSE-PHOSPHATE ISOMERASE WITH MODIFIED SUBSTRATE BINDING SITE | ||||||
 Components Components | TRIOSEPHOSPHATE ISOMERASE | ||||||
 Keywords Keywords | ISOMERASE / TIM BARREL / MODIFIED LOOP-8 | ||||||
| Function / homology |  Function and homology informationglycosome / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytosol / cytoplasm Function and homology informationglycosome / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Trypanosoma brucei brucei (eukaryote) Trypanosoma brucei brucei (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 2.65 Å | ||||||
 Authors Authors | Norledge, B.V. / Lambeir, A.M. / Abagyan, R.A. / Rottman, A. / Fernandez, A.M. / Filimonov, V.V. / Peter, M.G. / Wierenga, R.K. | ||||||
 Citation Citation | Journal: Proteins / Year: 2001 Title: Modeling, mutagenesis, and structural studies on the fully conserved phosphate-binding loop (loop 8) of triosephosphate isomerase: toward a new substrate specificity. Authors: Norledge, B.V. / Lambeir, A.M. / Abagyan, R.A. / Rottmann, A. / Fernandez, A.M. / Filimonov, V.V. / Peter, M.G. / Wierenga, R.K. #1: Journal: Structure / Year: 1993Title: The Crystal Structure of an Engineered Monomeric Triosephosphate Isomerase, monoTIM: The Correct Modelling of an Eight-Residue Loop. Authors: Borchert, T.V. / Abagyan, R. / Radha Kishan, K.V. / Zeelen, J.P. / Wierenga, R.K. #2: Journal: Protein Eng. / Year: 1997Title: Protein Engineering with Monomeric Triosephosphate Isomerase (monoTIM): The Modelling and Structure Verification of a Seven Residue Loop Authors: Thanki, N. / Zeelen, J.P. / Mathieu, M. / Jaenicke, R. / Abagyan, R.A. / Wierenga, R.K. / Schliebs, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dkw.cif.gz | 102.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dkw.ent.gz | 79.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1dkw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dk/1dkwftp://data.pdbj.org/pub/pdb/validation_reports/dk/1dkw | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25787.414 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Trypanosoma brucei brucei (eukaryote) / Species: Trypanosoma brucei / Strain: brucei / Production host:  Escherichia coli (E. coli) / References: UniProt: P04789, triose-phosphate isomerase Escherichia coli (E. coli) / References: UniProt: P04789, triose-phosphate isomerase#2: Chemical | Tert-Butyl alcohol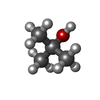  Mass: 74.122 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O Mass: 74.122 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 145 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 145 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.8 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 1.0 M CITRIC ACID PH 6.5, 20% PEG6000, 2.5% T-BUTANOL, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K | |||||||||||||||
| Crystal grow | *PLUS Method: unknown | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR571 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 20, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→22 Å / Num. all: 12311 / Num. obs: 12311 / % possible obs: 92.9 % / Observed criterion σ(I): -3 / Redundancy: 3.6 % / Biso Wilson estimate: 40 Å2 / Rmerge(I) obs: 0.078 / Net I/σ(I): 8.7 |
| Reflection shell | Resolution: 2.65→2.74 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.181 / % possible all: 73.4 |
| Reflection | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 22 Å / Num. obs: 13258 / % possible obs: 92.9 % / Observed criterion σ(I): -3 / Redundancy: 3.6 % / Rmerge(I) obs: 0.087 / Biso Wilson estimate: 40 Å2 |
| Reflection shell | *PLUS Highest resolution: 2.65 Å / Lowest resolution: 2.74 Å / % possible obs: 73.4 % / Redundancy: 2.4 % / Rmerge(I) obs: 0.181 / Mean I/σ(I) obs: 3.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.65→22 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→22 Å
| |||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.65 Å / Lowest resolution: 22 Å / Num. reflection all: 12114 / Num. reflection obs: 12114 / σ(I): 0 / σ(F): 0 / Num. reflection Rfree: 593 / % reflection Rfree: 5 % / Rfactor Rfree: 0.242 / Rfactor Rwork: 0.183 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 49.1 Å2 | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_bond_d / Dev ideal: 0.013 |
