- PDB-2wys: High resolution crystallographic structure of the Clostridium the... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 2wys
タイトル
High resolution crystallographic structure of the Clostridium thermocellum N-terminal endo-1,4-beta-D-xylanase 10B (Xyn10B) CBM22-1- GH10 modules complexed with xylohexaose
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AF" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AF" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BE" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
ENGINEERED RESIDUE IN CHAIN A, GLU 337 TO ALA ENGINEERED RESIDUE IN CHAIN B, GLU 337 TO ALA
非ポリマーの詳細
POTASSIUM (K): FROM THE CRYSTALLIZATION BUFFER PHOSPHATE ION (PO4): FRO THE CRYSTALLIZATION BUFFER ...POTASSIUM (K): FROM THE CRYSTALLIZATION BUFFER PHOSPHATE ION (PO4): FRO THE CRYSTALLIZATION BUFFER 2 XYLOHEXOSES (BUT ONLY 3 OF THE 6 XYLOSE RINGS CAN BE SEEN IN THE ELECTRON DENSITY), 2 PHOSPHATE AND 4 POTASSIUM IONS.
配列の詳細
THE PROTEIN SEQUENCE OF THE CONSTRUCT CORRESPONDS TO AMINO ACID RESIDUES 32 TO 551 WITH THE E337A ...THE PROTEIN SEQUENCE OF THE CONSTRUCT CORRESPONDS TO AMINO ACID RESIDUES 32 TO 551 WITH THE E337A MUTATION AND AN ADDITIONAL TWENTY AMINO ACID RESIDUES AT THE N-TERMINUS, MGSSHHHHHHSSGLVPRGSH. LINKER REGION BETWEEN AMINO ACID RESIDUES A182 TO A188 AND B182 AND B185 ARE NOT SEEN IN THE ELECTRON DENSITY MAPS.
-
実験情報
-
実験
実験
手法: X線回折 / 使用した結晶の数: 1
-
試料調製
結晶
マシュー密度: 4.9 Å3/Da / 溶媒含有率: 75 % 解説: PEAK DATA WAS THE BEST DATA COLLECTED OVER THE THREE WAVELENGTHS AND WAS USED FOR MODELING THE STRUCTURE
解像度: 2.75→100.5 Å / Num. obs: 61428 / % possible obs: 100 % / Observed criterion σ(I): 1.4 / 冗長度: 9.6 % / Biso Wilson estimate: 71.1 Å2 / Rmerge(I) obs: 0.19 / Net I/σ(I): 13
反射 シェル
解像度: 2.75→2.8 Å / 冗長度: 9.1 % / Rmerge(I) obs: 1 / Mean I/σ(I) obs: 1.4 / % possible all: 99.9
-
解析
ソフトウェア
名称
バージョン
分類
REFMAC
5.5.0102
精密化
MOSFLM
データ削減
SCALA
データスケーリング
SHELX
SOLVERESOLVE
位相決定
精密化
構造決定の手法: 多波長異常分散 開始モデル: NONE 解像度: 2.75→86.85 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.921 / SU B: 10.849 / SU ML: 0.205 / 交差検証法: THROUGHOUT / ESU R: 0.309 / ESU R Free: 0.253 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY.
Rfactor
反射数
%反射
Selection details
Rfree
0.23629
3102
5.1 %
RANDOM
Rwork
0.18516
-
-
-
obs
0.18771
58230
99.84 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード HYDROLASE (加水分解酵素) / XYLAN DEGRADATION /
HYDROLASE (加水分解酵素) / XYLAN DEGRADATION /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード





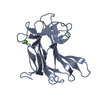















 PDBj
PDBj
 集合体
集合体


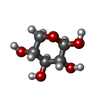


 タイプ: D-saccharide, beta linking / 分子量: 150.130 Da / 分子数: 1 / 由来タイプ: 組換発現 / 式: C5H10O5
タイプ: D-saccharide, beta linking / 分子量: 150.130 Da / 分子数: 1 / 由来タイプ: 組換発現 / 式: C5H10O5


 分子量: 40.078 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Ca
分子量: 40.078 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Ca 分子量: 94.971 Da / 分子数: 2 / 由来タイプ: 合成 / 式: PO4
分子量: 94.971 Da / 分子数: 2 / 由来タイプ: 合成 / 式: PO4 試料調製
試料調製 / ビームライン: ID29 / 波長: 0.9791, 0.9793, 0.9756
/ ビームライン: ID29 / 波長: 0.9791, 0.9793, 0.9756 解析
解析