 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2hy3: Crystal structure of the human tyrosine receptor phosphate gamma ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2hy3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human tyrosine receptor phosphate gamma in complex with vanadate | ||||||
 Components Components | Receptor-type tyrosine-protein phosphatase gamma | ||||||
 Keywords Keywords |  HYDROLASE / twisted mixed beta-sheets flanked by {alpha}-helices / Structural Genomics / PSI / Protein Structure Initiative / New York SGX Research Center for Structural Genomics / NYSGXRC HYDROLASE / twisted mixed beta-sheets flanked by {alpha}-helices / Structural Genomics / PSI / Protein Structure Initiative / New York SGX Research Center for Structural Genomics / NYSGXRC | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of epithelial cell migration / transmembrane receptor protein tyrosine phosphatase activity / dephosphorylation / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / cell surface receptor protein tyrosine kinase signaling pathway / negative regulation of neuron projection development / extracellular exosome / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.6 Å | ||||||
 Authors Authors | Jin, X. / Min, T. / Bera, A. / Mu, H. / Sauder, J.M. / Freeman, J.C. / Reyes, C. / Smith, D. / Wasserman, S.R. / Burley, S.K. ...Jin, X. / Min, T. / Bera, A. / Mu, H. / Sauder, J.M. / Freeman, J.C. / Reyes, C. / Smith, D. / Wasserman, S.R. / Burley, S.K. / Shapiro, L. / New York SGX Research Center for Structural Genomics (NYSGXRC) | ||||||
 Citation Citation | Journal: J.STRUCT.FUNCT.GENOM. / Year: 2007 Title: Structural genomics of protein phosphatases. Authors: Almo, S.C. / Bonanno, J.B. / Sauder, J.M. / Emtage, S. / Dilorenzo, T.P. / Malashkevich, V. / Wasserman, S.R. / Swaminathan, S. / Eswaramoorthy, S. / Agarwal, R. / Kumaran, D. / Madegowda, M. ...Authors: Almo, S.C. / Bonanno, J.B. / Sauder, J.M. / Emtage, S. / Dilorenzo, T.P. / Malashkevich, V. / Wasserman, S.R. / Swaminathan, S. / Eswaramoorthy, S. / Agarwal, R. / Kumaran, D. / Madegowda, M. / Ragumani, S. / Patskovsky, Y. / Alvarado, J. / Ramagopal, U.A. / Faber-Barata, J. / Chance, M.R. / Sali, A. / Fiser, A. / Zhang, Z.Y. / Lawrence, D.S. / Burley, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2hy3.cif.gz | 121.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2hy3.ent.gz | 100 KB | Display | PDB format |
| PDBx/mmJSON format | 2hy3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hy/2hy3ftp://data.pdbj.org/pub/pdb/validation_reports/hy/2hy3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 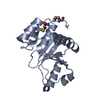 1rxdC  2fh7C  2g59C  2hcmC  2hhlC  2hxpC  2i0oC  2i1yC  2i44C  2iq1C  2irmC  2isnC  2nv5C  2oycC  2p27C  2p4uC  2p69C  2p8eC  2pbnC  2q5eC  2qjcC  2r0bC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg label comp-ID: GLU / End label comp-ID: GLY / Refine code: 4 / Auth seq-ID: 826 - 1122 / Label seq-ID: 9 - 305
|
-Components
| #1: Protein | Mass: 36417.207 Da / Num. of mol.: 2 / Fragment: residues 819-1130 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PTPRG / Plasmid: pSGX4 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P23470, protein-tyrosine-phosphatase Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P23470, protein-tyrosine-phosphatase#2: Chemical | Vanadate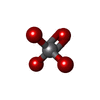  Mass: 114.939 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: VO4 Mass: 114.939 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: VO4#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 50 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 50 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.09 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 12% PEG3350, 0.1M Tris, 0.3M NaCl, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Wavelength: 0.97891 Å / Beamline: X4A / Wavelength: 0.97891 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jul 16, 2006 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97891 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→50 Å / Num. all: 24687 / Num. obs: 20061 / % possible obs: 87 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 2 |
| Reflection shell | Resolution: 2.6→2.69 Å / % possible all: 89 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.6→17 Å / Cor.coef. Fo:Fc: 0.94 / Cor.coef. Fo:Fc free: 0.882 / SU B: 25.526 / SU ML: 0.265 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.426 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: Used weighted full matrix least squares procedure.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.551 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→17 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 2217 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.666 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
