 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2hhl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human small CTD phosphatase 3 isoform 1 | ||||||
 Components Components | CTD small phosphatase-like protein | ||||||
 Keywords Keywords |  HYDROLASE / CTD phosphatase / Keggins anion / Structural Genomics / PSI / Protein Structure Initiative / New York SGX Research Center for Structural Genomics / NYSGXRC HYDROLASE / CTD phosphatase / Keggins anion / Structural Genomics / PSI / Protein Structure Initiative / New York SGX Research Center for Structural Genomics / NYSGXRC | ||||||
| Function / homology |  Function and homology information Function and homology informationRNA polymerase II CTD heptapeptide repeat phosphatase activity / negative regulation of G1/S transition of mitotic cell cycle / protein-serine/threonine phosphatase / phosphoprotein phosphatase activity / negative regulation of protein phosphorylation / extracellular exosome / metal ion binding / nucleusSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Malashkevich, V.N. / Toro, R. / Ramagopal, U. / Sauder, J.M. / Schwinn, K.D. / Thompson, D.A. / Rutter, M.E. / Dickey, M. / Groshong, C. / Bain, K.T. ...Malashkevich, V.N. / Toro, R. / Ramagopal, U. / Sauder, J.M. / Schwinn, K.D. / Thompson, D.A. / Rutter, M.E. / Dickey, M. / Groshong, C. / Bain, K.T. / Adams, J.M. / Reyes, C. / Rooney, I. / Powell, A. / Boice, A. / Gheyi, T. / Ozyurt, S. / Atwell, S. / Wasserman, S.R. / Emtage, S. / Burley, S.K. / Almo, S.C. / New York SGX Research Center for Structural Genomics (NYSGXRC) | ||||||
 Citation Citation | Journal: J.STRUCT.FUNCT.GENOM. / Year: 2007 Title: Structural genomics of protein phosphatases. Authors: Almo, S.C. / Bonanno, J.B. / Sauder, J.M. / Emtage, S. / Dilorenzo, T.P. / Malashkevich, V. / Wasserman, S.R. / Swaminathan, S. / Eswaramoorthy, S. / Agarwal, R. / Kumaran, D. / Madegowda, M. ...Authors: Almo, S.C. / Bonanno, J.B. / Sauder, J.M. / Emtage, S. / Dilorenzo, T.P. / Malashkevich, V. / Wasserman, S.R. / Swaminathan, S. / Eswaramoorthy, S. / Agarwal, R. / Kumaran, D. / Madegowda, M. / Ragumani, S. / Patskovsky, Y. / Alvarado, J. / Ramagopal, U.A. / Faber-Barata, J. / Chance, M.R. / Sali, A. / Fiser, A. / Zhang, Z.Y. / Lawrence, D.S. / Burley, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2hhl.cif.gz | 170.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2hhl.ent.gz | 134.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2hhl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hh/2hhlftp://data.pdbj.org/pub/pdb/validation_reports/hh/2hhl | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 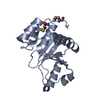 1rxdC  2fh7C  2g59C  2hcmC  2hxpC  2hy3C  2i0oC  2i1yC  2i44C  2iq1C  2irmC  2isnC  2nv5C  2oycC  2p27C  2p4uC  2p69C  2p8eC  2pbnC  2q5eC  2qjcC  2r0bC  1ta0S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22424.609 Da / Num. of mol.: 4 / Fragment: residues 82-265 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTDSPL, C3orf8, NIF1, NIFL, YA22 / Production host:  Escherichia coli (E. coli) / References: UniProt: O15194 Escherichia coli (E. coli) / References: UniProt: O15194#2: Chemical | 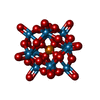  Mass: 2877.030 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: O40PW12 Mass: 2877.030 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: O40PW12#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 540 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 540 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.35 Å3/Da / Density % sol: 47.66 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 5.5 Details: 0.2M lithium sulfate monohydrate, 0.1M bis-tris, 25% w/v PEG 3350, pH 5.5, VAPOR DIFFUSION, SITTING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X6A / Wavelength: 1.74 Å / Beamline: X6A / Wavelength: 1.74 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 21, 2006 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: X6A / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.74 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 1.7 % / Av σ(I) over netI: 8.5 / Number: 139989 / Rmerge(I) obs: 0.056 / Χ2: 1.05 / D res high: 2.1 Å / D res low: 50 Å / Num. obs: 84087 / % possible obs: 87.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→20 Å / Num. obs: 47666 / % possible obs: 87.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 1.7 % / Biso Wilson estimate: 26.5 Å2 / Rmerge(I) obs: 0.056 / Rsym value: 0.056 / Χ2: 1.048 / Net I/σ(I): 8.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 1.4 % / Rmerge(I) obs: 0.265 / Num. unique all: 7128 / Χ2: 0.974 / % possible all: 74 |
-Phasing
| Phasing MR | Rfactor: 0.499 / Cor.coef. Fo:Fc: 0.509
|
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TA0 Resolution: 2.1→19.86 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.915 / SU B: 4.986 / SU ML: 0.135 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.237 / ESU R Free: 0.206 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.238 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→19.86 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.154 Å / Total num. of bins used: 20
|
