 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1yqc: Crystal Structure of Ureidoglycolate Hydrolase (AllA) from Escher... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1yqc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Ureidoglycolate Hydrolase (AllA) from Escherichia coli O157:H7 | ||||||
 Components Components | Ureidoglycolate hydrolase | ||||||
 Keywords Keywords | HYDROLASE / AllA / Ureidoglycolate / Purine metabolism / Structural Genomics / Montreal-Kingston Bacterial Structural Genomics Initiative / BSGI | ||||||
| Function / homology |  Function and homology informationureidoglycolate lyase / ureidoglycolate lyase activity / ureidoglycolate hydrolase activity / allantoin catabolic process / purine nucleobase catabolic process Function and homology informationureidoglycolate lyase / ureidoglycolate lyase activity / ureidoglycolate hydrolase activity / allantoin catabolic process / purine nucleobase catabolic processSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.709 Å | ||||||
 Authors Authors | Raymond, S. / Tocilj, A. / Matte, A. / Cygler, M. / Montreal-Kingston Bacterial Structural Genomics Initiative (BSGI) | ||||||
 Citation Citation | Journal: Proteins / Year: 2005 Title: Crystal structure of ureidoglycolate hydrolase (AllA) from Escherichia coli O157:H7 Authors: Raymond, S. / Tocilj, A. / Ajamian, E. / Li, Y. / Hung, M.-N. / Matte, A. / Cygler, M. #1: Journal: METHODS ENZYMOL. / Year: 1997 Title: Processing of X-ray Diffraction Data Collected in Oscillation Mode Authors: Otwinowski, Z. / Minor, W. #2: Journal: ACTA CRYSTALLOGR.,SECT.D / Year: 1999 Title: Automated MAD and MIR structure solution Authors: Terwilliger, T.C. / Berendzen, J. #3: Journal: ACTA CRYSTALLOGR.,SECT.D / Year: 2000 Title: Maximum-likelihood density modification Authors: Terwilliger, T.C. #4: Journal: ACTA CRYSTALLOGR.,SECT.D / Year: 1997 Title: Refinement of macromolecular structures by the maximum-likelihood method Authors: Murshudov, G.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1yqc.cif.gz | 83 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1yqc.ent.gz | 65 KB | Display | PDB format |
| PDBx/mmJSON format | 1yqc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yq/1yqcftp://data.pdbj.org/pub/pdb/validation_reports/yq/1yqc | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 19584.639 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Strain: O157:H7 / Gene: AllA / Plasmid: pET20b / Production host: Escherichia coli (E. coli) / References: UniProt: P63486, EC: 3.5.3.19#2: Chemical | ChemComp-GLV / | Glyoxylic acid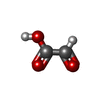  Mass: 74.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H2O3 Mass: 74.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H2O3#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 319 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 319 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43 % |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.9611, 0.9800, 0.9802 / Beamline: X8C / Wavelength: 0.9611, 0.9800, 0.9802 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Oct 2, 2004 / Details: Mirrors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Mirrors / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.71→50 Å / Num. obs: 36248 / % possible obs: 96.2 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing set |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MAD set |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set site |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm | FOM : 0.67 / FOM acentric: 0.67 / FOM centric: 0.7 / Reflection: 22052 / Reflection acentric: 21213 / Reflection centric: 839 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.709→36.442 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.925 / WRfactor Rfree: 0.267 / WRfactor Rwork: 0.223 / TLS residual ADP flag: LIKELY RESIDUAL / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1.71 / ESU R: 0.143 / ESU R Free: 0.138 / Stereochemistry target values: Engh & Huber / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.234 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.709→36.442 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Refine-ID: X-RAY DIFFRACTION / Selection: ALL / Auth seq-ID: 1 - 160 / Label seq-ID: 1 - 160
|
