 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jnu: Photoexcited structure of the plant photoreceptor domain, phy3 LOV2 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jnu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Photoexcited structure of the plant photoreceptor domain, phy3 LOV2 | ||||||
 Components Components | PHY3 PROTEIN | ||||||
 Keywords Keywords |  SIGNALING PROTEIN / ELECTRON TRANSPORT / cysteinyl-flavin adduct / photoexcited / PAS / LOV / plant photoreceptor / phototropin / photochemistry / light-driven bond / phy3 SIGNALING PROTEIN / ELECTRON TRANSPORT / cysteinyl-flavin adduct / photoexcited / PAS / LOV / plant photoreceptor / phototropin / photochemistry / light-driven bond / phy3 | ||||||
| Function / homology |  Function and homology information Function and homology informationblue light photoreceptor activity / detection of visible light / non-specific serine/threonine protein kinase / protein kinase activity / phosphorylation / regulation of DNA-templated transcription / ATP bindingSimilarity search - Function | ||||||
| Biological species |  Adiantum capillus-veneris (maidenhair fern) Adiantum capillus-veneris (maidenhair fern) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Crosson, S. / Moffat, K. | ||||||
 Citation Citation | Journal: Plant Cell / Year: 2002 Title: Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Authors: Crosson, S. / Moffat, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jnu.cif.gz | 97.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jnu.ent.gz | 74.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1jnu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jn/1jnuftp://data.pdbj.org/pub/pdb/validation_reports/jn/1jnu | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| 3 | 
| ||||||||||
| 4 | 
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 12181.764 Da / Num. of mol.: 4 Fragment: FMN-BINDING DOMAIN OF CHIMERIC PHYTOCHROME/PHOTOTROPIN PHOTORECEPTOR, LOV3 Source method: isolated from a genetically manipulated source Details: contains a cysteinyl-flavin C(4a) covalent adduct Source: (gene. exp.) Adiantum capillus-veneris (maidenhair fern)Gene: phy3 / Plasmid: pcal-n-ek / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21-DE3 / References: UniProt: Q9ZWQ6 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21-DE3 / References: UniProt: Q9ZWQ6#2: Chemical | ChemComp-FMN / Flavin mononucleotide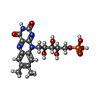  Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 65 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 65 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.56 Å3/Da / Density % sol: 65.47 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PEG 5000 MME, Tris, Glycerol, pH 8.0, VAPOR DIFFUSION, HANGING DROP at 293K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 25 ℃ / Method: unknown | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 296 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 1 Å / Beamline: 14-BM-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 5, 2001 |
| Radiation | Monochromator: Silicon / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→26.8 Å / Num. all: 29191 / Num. obs: 17700 / % possible obs: 87.5 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 1 / Redundancy: 1.65 % / Rmerge(I) obs: 0.044 / Net I/σ(I): 12.3 |
| Reflection shell | Resolution: 2.6→2.69 Å / Rmerge(I) obs: 0.19 / Mean I/σ(I) obs: 3.2 / Num. unique all: 1583 / % possible all: 79 |
| Reflection | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 30 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.044 |
| Reflection shell | *PLUS % possible obs: 79 % / Rmerge(I) obs: 0.19 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: phy3 LOV2 dark-state structure Resolution: 2.6→26.8 Å / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.69 Å2
| ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→26.8 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 30 Å / % reflection Rfree: 7.3 % / Rfactor obs: 0.238 / Rfactor Rfree: 0.259 / Rfactor Rwork: 0.238 | ||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 32.7 Å2 | ||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
