 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1dzo | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Truncated PAK pilin from Pseudomonas aeruginosa | ||||||
 要素 要素 | TYPE IV PILIN | ||||||
 キーワード キーワード |  CELL ADHESION (細胞接着) / LECTIN (レクチン) / ADHESIN CELL ADHESION (細胞接着) / LECTIN (レクチン) / ADHESIN | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |  PSEUDOMONAS AERUGINOSA PAK (緑膿菌) PSEUDOMONAS AERUGINOSA PAK (緑膿菌) | ||||||
| 手法 | X線回折 / 多重同系置換・異常分散 / 解像度: 1.63 Å | ||||||
 データ登録者 データ登録者 | Hazes, B. / Read, R.J. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 2000 タイトル: Crystal Structure of Pseudomonas Aeruginosa Pak Pilin Suggests a Main-Chain-Dominated Mode of Receptor Binding 著者: Hazes, B. / Sastry, P.A. / Hayakawa, K. / Read, R.J. / Irvin, R.T. | ||||||
| 履歴 |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: DSSP |
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1dzo.cif.gz | 57.4 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1dzo.ent.gz | 45.3 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1dzo.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/dz/1dzoftp://data.pdbj.org/pub/pdb/validation_reports/dz/1dzo | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ | |
|---|---|
| 類似構造データ |












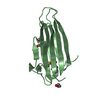
-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | ON CELLS PILIN IS FOUND AS LONG THIN FIBERS WHICH MEDIATE CELL ATTACHMENT. BASED ON MOLECULAR MODELING A PRELIMINARY FIBER MODEL HAS BEEN PROPOSED FOR THE RELATED TYPE IV PILIN OF NEISSERIA GONORRHOEAE(PDB ID CODE 1AY2). TO GENERATE THE CORRESPONDING MODEL FOR PAK PILIN THE COORDINATES IN THIS ENTRY SHOULD BE SUPERIMPOSED ON THE NEISSERIA MODEL FOLLOWED BY THE APPLICATION OF THE TRANSFORMATIONS AS INDICATED IN PDB ENTRY 1AY2.PDB ALTHOUGH THE NEISSERIA MODEL IS THE BEST CURRENT MODEL FOR THE FIBER STRUCTURE, IT SHOULD BE KEPT IN MIND THAT SIGNIFICANT DEVIATIONS FROM REALITY MAY EXIST. IN PARTICULAR, IT MAY BE POSSIBLE TO CREATE A SIMILAR MODEL BY STACKING PERFECT PENTAMERS OF PILIN MOLECULES. THE TYPE IV PILUS IS POLAR AND IT APPEARS TO EXPOSE EXTREMELY HYDROPHOBIC ALPHA HELICES AT ONE OF ITS ENDS. BASED ON RECEPTOR BINDING CONSIDERATIONS WE HAVE PROPOSED THAT THE HYDROPHOBIC ALPHA HELICES ARE DISPLAYED AT THE TIP OF THE PILUS AND THEREFORE INTERACT WITH HOST CELLS. THIS CONTRASTS WITH EARLIER MODELS WHERE THE HELICES WERE ASSUMED TO BE BURIED IN THE BACTERIAL OUTER MEMBRANE. |
-要素
| #1: タンパク質 | 分子量: 12696.225 Da / 分子数: 1 / 断片: GLOBULAR DOMAIN / 変異: YES / 由来タイプ: 組換発現 由来: (組換発現) PSEUDOMONAS AERUGINOSA PAK (緑膿菌)解説: RESIDUES 22-28 ARE FROM THE EXPRESSION VECTOR. RESIDUES 29-144 ARE FROM THE MATURE PROTEIN. 細胞内の位置: EXTRACELLULAR FILAMENTOUS APPENDAGE / 遺伝子: PILA / プラスミド: PRLD / 細胞内の位置 (発現宿主): PERIPLASMIC SPACE / 発現宿主: ESCHERICHIA COLI (大腸菌) / 株 (発現宿主): BL21 / 参照: UniProt: P02973 |
|---|---|
| #2: 水 | ChemComp-HOH / 水 分子量: 18.015 Da / 分子数: 131 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 131 / 由来タイプ: 天然 / 式: H2O |
| 構成要素の詳細 | CHAIN A IS A DELETION MUTANT, MISSING RESIDUES 1-28 OF THE NATIVE SEQUENCE. THE RECOMBINANT PROTEIN ...CHAIN A IS A DELETION MUTANT, MISSING RESIDUES 1-28 OF THE NATIVE SEQUENCE. THE RECOMBINAN |
| 配列の詳細 | RESIDUES 22-28 ARE FROM THE EXPRESSION VECTOR. RESIDUES 22-24 HAVE NOT BEEN MODELED DUE TO LACK OF ...RESIDUES 22-28 ARE FROM THE EXPRESSION |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.14 Å3/Da / 溶媒含有率: 43 % 解説: DERIVATIVE DATA WERE SCALED USING THE NATIVE DATA AS A REFERENCE | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 8.2 詳細: HANGING DROP USING 1 ML OF RESERVOIR DROPS MADE FROM 3 MICROLITRE PROTEIN AND 3 MICROLITRE OF MOTHER LIQUOR PROTEIN SOLUTION = 10 MG/ML IN WATER MOTHER LIQUOR = 60% (NH4)2SO4, 0.1M HEPES PH 8.2 | ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 手法: unknown | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 293 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: ELLIOTT GX-13 / 波長: 1.5418 |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 2000年12月15日 / 詳細: SUPPER MIRROR |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 1.633→37.44 Å / Num. obs: 253714 / % possible obs: 99.5 % / 冗長度: 7.6 % / Biso Wilson estimate: 16.8 Å2 / Rsym value: 0.049 / Net I/σ(I): 26.7 |
| 反射 シェル | 解像度: 1.63→1.72 Å / 冗長度: 6.6 % / Mean I/σ(I) obs: 10.1 / Rsym value: 0.19 / % possible all: 96.4 |
| 反射 | *PLUS Num. obs: 14500 / Num. measured all: 110628 / Rmerge(I) obs: 0.049 |
| 反射 シェル | *PLUS % possible obs: 96.4 % / Rmerge(I) obs: 0.19 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多重同系置換・異常分散 / 解像度: 1.63→37.44 Å / SU B: 1.26 / SU ML: 0.04 / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.12 / ESU R Free: 0.08 詳細: CNS EXPLICIT BULK SOLVENT CORRECTION WAS USED. B-SPHERE RMS = 1.851 FOR FREE ATOMS AND 2.429 FOR BONDED ATOMS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 16.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.63→37.44 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
|
