 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b8d | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A PHYCOUROBILIN-CONTAINING PHYCOERYTHRIN | ||||||
 Components Components | (PROTEIN (RHODOPHYTAN PHYCOERYTHRIN ...) x 3 | ||||||
 Keywords Keywords |  PHOTOSYNTHESIS / LIGHT-HARVESTING COMPLEX / RED ALGAE / PHYCOBILIPROTEIN PHOTOSYNTHESIS / LIGHT-HARVESTING COMPLEX / RED ALGAE / PHYCOBILIPROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Griffithsia monilis (eukaryote) Griffithsia monilis (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Ritter, S. / Hiller, R.G. / Wrench, P.M. / Welte, W. / Diederichs, K. | ||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 1999 Title: Crystal structure of a phycourobilin-containing phycoerythrin at 1.90-A resolution. Authors: Ritter, S. / Hiller, R.G. / Wrench, P.M. / Welte, W. / Diederichs, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b8d.cif.gz | 193.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b8d.ent.gz | 156 KB | Display | PDB format |
| PDBx/mmJSON format | 1b8d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b8/1b8dftp://data.pdbj.org/pub/pdb/validation_reports/b8/1b8d | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1cpcS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS oper:
|
-Components
-PROTEIN (RHODOPHYTAN PHYCOERYTHRIN ... , 3 types, 5 molecules AKBLG
| #1: Protein | Mass: 17687.834 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: THE PHYCOBILISOMES ARE LOCATED ON THE STROMAL SIDE OF THE THYLAKOID MEMBRANES. Source: (natural) Griffithsia monilis (eukaryote) / Organelle: RHODOPLAST / References: UniProt: O36005#2: Protein | Mass: 18511.941 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: THE PHYCOBILISOMES ARE LOCATED ON THE STROMAL SIDE OF THE THYLAKOID MEMBRANES. Source: (natural) Griffithsia monilis (eukaryote) / Organelle: RHODOPLAST / References: UniProt: O36004#3: Protein/peptide | | Mass: 656.729 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Griffithsia monilis (eukaryote) / Organelle: RHODOPLAST |
|---|
-Non-polymers , 3 types, 343 molecules 
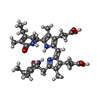

| #4: Chemical | ChemComp-PEB / Phycoerythrobilin Mass: 588.694 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C33H40N4O6 Mass: 588.694 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C33H40N4O6#5: Chemical | Phycourobilin Mass: 590.710 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H42N4O6 Mass: 590.710 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H42N4O6#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 51 % |
|---|---|
| Crystal grow | pH: 7.5 Details: 700MM SODIUM ACETATE,5MM KCL,100MM IMIDAZOLE, PH 7.5 AT 17C |
| Crystal grow | *PLUS Method: otherDetails: .refer to Ritter, S., (1997) Protein Peptide Lett., 4, 69. |
-Data collection
| Diffraction | Mean temperature: 289 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.92 / Beamline: X11 / Wavelength: 0.92 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 1, 1993 |
| Radiation | Monochromator: SI (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 1.82→15 Å / Num. obs: 67598 / % possible obs: 97 % / Observed criterion σ(I): 0 / Redundancy: 1.9 % / Biso Wilson estimate: 18 Å2 / Rmerge(I) obs: 0.059 |
| Reflection shell | Resolution: 1.82→2 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.313 / % possible all: 92 |
| Reflection | *PLUS % possible obs: 96.8 % / Num. measured all: 131195 |
| Reflection shell | *PLUS % possible obs: 92 % / Num. unique obs: 15857 / Num. measured obs: 28679 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1CPC Resolution: 1.9→100 Å / Data cutoff low absF: 0.001 / Cross valid method: THROUGHOUT / σ(F): 0 / Details: INDIVIDUAL B-FAC REFINEMENT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.15 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.27 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Weight Biso : 2 / Weight position: 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.97 Å / Total num. of bins used: 10 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
