 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eyx | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF R-PHYCOERYTHRIN AT 2.2 ANGSTROMS | ||||||
 Components Components | (R-PHYCOERYTHRIN) x 3 | ||||||
 Keywords Keywords |  PHOTOSYNTHESIS / R-PHYCOERYTHRIN / MACROSEEDING / TWIN / PROTEIN STRUCTURE / SEQUENCES / PHYCOBILIPROTEIN / RED ALGAE PHOTOSYNTHESIS / R-PHYCOERYTHRIN / MACROSEEDING / TWIN / PROTEIN STRUCTURE / SEQUENCES / PHYCOBILIPROTEIN / RED ALGAE | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Gracilaria chilensis (eukaryote) Gracilaria chilensis (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Contreras-Martel, C. / Legrand, P. / Piras, C. / Vernede, X. / Martinez-Oyanedel, J. / Bunster, M. / Fontecilla-Camps, J.C. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Crystallization and 2.2 A resolution structure of R-phycoerythrin from Gracilaria chilensis: a case of perfect hemihedral twinning. Authors: Contreras-Martel, C. / Martinez-Oyanedel, J. / Bunster, M. / Legrand, P. / Piras, C. / Vernede, X. / Fontecilla-Camps, J.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eyx.cif.gz | 155.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eyx.ent.gz | 123.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1eyx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ey/1eyxftp://data.pdbj.org/pub/pdb/validation_reports/ey/1eyx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1liaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 4 molecules AKBL
| #1: Protein | Mass: 17770.857 Da / Num. of mol.: 2 / Fragment: ALPHA CHAIN / Source method: isolated from a natural source / Source: (natural) Gracilaria chilensis (eukaryote) / Organelle: CHLOROPLAST / References: UniProt: Q7SIG0#2: Protein | Mass: 18634.182 Da / Num. of mol.: 2 / Fragment: BETA CHAIN / Source method: isolated from a natural source / Source: (natural) Gracilaria chilensis (eukaryote) / Organelle: CHLOROPLAST / References: UniProt: Q7SIF9 |
|---|
-Protein/peptide , 1 types, 2 molecules GH
| #3: Protein/peptide | Mass: 606.694 Da / Num. of mol.: 2 / Fragment: GAMMA CHAIN / Source method: isolated from a natural source / Source: (natural) Gracilaria chilensis (eukaryote) / Organelle: CHLOROPLAST |
|---|
-Non-polymers , 5 types, 122 molecules 
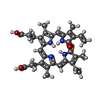
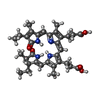
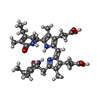

| #4: Chemical | Sulfate Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-BLA / |  Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6 Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6#6: Chemical | ChemComp-CYC / Phycocyanobilin Mass: 588.694 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C33H40N4O6 Mass: 588.694 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C33H40N4O6#7: Chemical | Phycourobilin Mass: 590.710 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H42N4O6 Mass: 590.710 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H42N4O6#8: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 110 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.88 Å3/Da / Density % sol: 34.7 % Description: A PERFECT HEMIHEDRAL TWINNED CRYSTAL. STANLEY DISTRIBUTION, 1.44. TWIN FRACTION, 0.48. | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 200 MM HEPES, 500 MM SODIUM CHLORIDE, 50 MM POTASIUM CHLORIDE, 15 MM SODIUM AZIDE, 16% AMMONIUM SULPHATE, pH 7.0, VAPOR DIFFUSION, SITTING DROP, temperature 293K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 294 K | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.9783 / Beamline: BM14 / Wavelength: 0.9783 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 31, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9783 Å / Relative weight: 1 |
| Reflection | Resolution: 2.16→42.55 Å / Num. obs: 38751 / % possible obs: 98.7 % / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Rmerge(I) obs: 0.101 / Rsym value: 0.101 / Net I/σ(I): 7.3 |
| Reflection shell | Resolution: 2.16→2.28 Å / Redundancy: 2 % / Rmerge(I) obs: 0.039 / Mean I/σ(I) obs: 2.8 / Rsym value: 0.039 / % possible all: 89.4 |
| Reflection | *PLUS Num. measured all: 123143 |
| Reflection shell | *PLUS % possible obs: 89.4 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1LIA Resolution: 2.25→999 Å / Num. parameters: 2290 / Num. restraintsaints: 3005 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 5654 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→999 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.18 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
