 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-24677 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of KIFBP core | ||||||||||||||||||
 Map data Map data | |||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology information central nervous system projection neuron axonogenesis / mitochondrial transport / kinesin binding / neuron projection maintenance / microtubule cytoskeleton organization / in utero embryonic development / cytoskeleton / mitochondrion central nervous system projection neuron axonogenesis / mitochondrial transport / kinesin binding / neuron projection maintenance / microtubule cytoskeleton organization / in utero embryonic development / cytoskeleton / mitochondrionSimilarity search - Function | ||||||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.8 Å | ||||||||||||||||||
 Authors Authors | Solon AL / Tan Z / Schutt KL / Jepsen L / Haynes SE / Nesvizhskii AI / Sept D / Stumpff J / Ohi R / Cianfrocco MA | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Sci Adv / Year: 2021 Title: Kinesin-binding protein remodels the kinesin motor to prevent microtubule binding. Authors: April L Solon / Zhenyu Tan / Katherine L Schutt / Lauren Jepsen / Sarah E Haynes / Alexey I Nesvizhskii / David Sept / Jason Stumpff / Ryoma Ohi / Michael A Cianfrocco / Abstract: Kinesins are regulated in space and time to ensure activation only in the presence of cargo. Kinesin-binding protein (KIFBP), which is mutated in Goldberg-Shprintzen syndrome, binds to and inhibits ...Kinesins are regulated in space and time to ensure activation only in the presence of cargo. Kinesin-binding protein (KIFBP), which is mutated in Goldberg-Shprintzen syndrome, binds to and inhibits the catalytic motor heads of 8 of 45 kinesin superfamily members, but the mechanism remains poorly defined. Here, we used cryo–electron microscopy and cross-linking mass spectrometry to determine high-resolution structures of KIFBP alone and in complex with two mitotic kinesins, revealing structural remodeling of kinesin by KIFBP. We find that KIFBP remodels kinesin motors and blocks microtubule binding (i) via allosteric changes to kinesin and (ii) by sterically blocking access to the microtubule. We identified two regions of KIFBP necessary for kinesin binding and cellular regulation during mitosis. Together, this work further elucidates the molecular mechanism of KIFBP-mediated kinesin inhibition and supports a model in which structural rearrangement of kinesin motor domains by KIFBP abrogates motor protein activity. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_24677.map.gz | 96.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-24677-v30.xmlemd-24677.xml | 18.8 KB 18.8 KB | Display Display | EMDB header |
| Images | 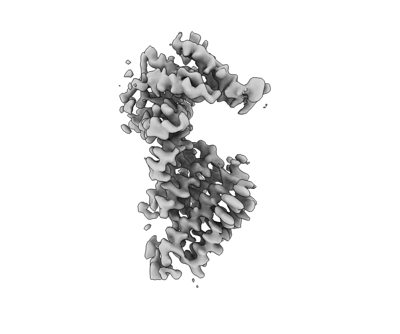 emd_24677.png emd_24677.png | 41.7 KB | ||
| Masks | emd_24677_msk_1.map | 103 MB | Mask map | |
| Others | emd_24677_additional_1.map.gzemd_24677_half_map_1.map.gzemd_24677_half_map_2.map.gz | 80.8 MB 80.8 MB 80.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-24677ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24677 http://ftp.pdbj.org/pub/emdb/structures/EMD-24677ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24677 | HTTPS FTP |
-Related structure data
| Related structure data |  7rsqMC  7rsiC  7rypC  7ryqC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_24677.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 0.98 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Mask #1
| File | emd_24677_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X








-Additional map: #1
| File | emd_24677_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |



-Half map: #1
| File | emd_24677_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |





-Half map: #2
| File | emd_24677_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : high-resolution structure of KIFBP(core)
| Entire | Name: high-resolution structure of KIFBP(core) |
|---|---|
| Components |
|
-Supramolecule #1: high-resolution structure of KIFBP(core)
| Supramolecule | Name: high-resolution structure of KIFBP(core) / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism:  Escherichia coli (E. coli) Escherichia coli (E. coli) |
-Macromolecule #1: KIF-binding protein
| Macromolecule | Name: KIF-binding protein / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 71.913945 KDa |
| Recombinant expression | Organism: Escherichia coli BL21 (bacteria) |
| Sequence | String: MANVPWAEVC EKFQAALALS RVELHKNPEK EPYKSKYSAR ALLEEVKALL GPAPEDEDER PEAEDGPGAG DHALGLPAEV VEPEGPVAQ RAVRLAVIEF HLGVNHIDTE ELSAGEEHLV KCLRLLRRYR LSHDCISLCI QAQNNLGILW SEREEIETAQ A YLESSEAL ...String: MANVPWAEVC EKFQAALALS RVELHKNPEK EPYKSKYSAR ALLEEVKALL GPAPEDEDER PEAEDGPGAG DHALGLPAEV VEPEGPVAQ RAVRLAVIEF HLGVNHIDTE ELSAGEEHLV KCLRLLRRYR LSHDCISLCI QAQNNLGILW SEREEIETAQ A YLESSEAL YNQYMKEVGS PPLDPTERFL PEEEKLTEQE RSKRFEKVYT HNLYYLAQVY QHLEMFEKAA HYCHSTLKRQ LE HNAYHPI EWAINAATLS QFYINKLCFM EARHCLSAAN VIFGQTGKIS ATEDTPEAEG EVPELYHQRK GEIARCWIKY CLT LMQNAQ LSMQDNIGEL DLDKQSELRA LRKKELDEEE SIRKKAVQFG TGELCDAISA VEEKVSYLRP LDFEEARELF LLGQ HYVFE AKEFFQIDGY VTDHIEVVQD HSALFKVLAF FETDMERRCK MHKRRIAMLE PLTVDLNPQY YLLVNRQIQF EIAHA YYDM MDLKVAIADR LRDPDSHIVK KINNLNKSAL KYYQLFLDSL RDPNKVFPEH IGEDVLRPAM LAKFRVARLY GKIITA DPK KELENLATSL EHYKFIVDYC EKHPEAAQEI EVELELSKEM VSLLPTKMER FRTKMALT |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.7 |
| Grid | Model: UltrAuFoil R1.2/1.3 / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS GLACIOS |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: BRIGHT FIELDBright-field microscopy |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Average electron dose: 60.0 e/Å2 |
-Image processing
| Particle selection | Number selected: 128190 |
|---|---|
| CTF correction | Software - Name: cryoSPARC |
| Startup model | Type of model: OTHER / Details: cryosparc ab-initio reconstruction |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION |
| Final reconstruction | Number classes used: 1 / Resolution.type: BY AUTHOR / Resolution: 3.8 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 128190 |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-7rsq: |
