 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6rqp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Steady-state-SMX dark state structure of bacteriorhodopsin | ||||||
 Components Components | Bacteriorhodopsin | ||||||
 Keywords Keywords | PROTON TRANSPORT / retinal / serial crystallography / time-resolved / TR-SMX | ||||||
| Function / homology |  Function and homology informationphotoreceptor activity / phototransduction / proton transmembrane transport / monoatomic ion channel activity / plasma membrane Function and homology informationphotoreceptor activity / phototransduction / proton transmembrane transport / monoatomic ion channel activity / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Halobacterium salinarum NRC-1 (Halophile) Halobacterium salinarum NRC-1 (Halophile) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.8 Å | ||||||
 Authors Authors | Weinert, T. / Skopintsev, P. / James, D. / Kekilli, D. / Furrer, A. / Bruenle, S. / Mous, S. / Nogly, P. / Standfuss, J. | ||||||
 Citation Citation | Journal: Science / Year: 2019 Title: Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Authors: Weinert, T. / Skopintsev, P. / James, D. / Dworkowski, F. / Panepucci, E. / Kekilli, D. / Furrer, A. / Brunle, S. / Mous, S. / Ozerov, D. / Nogly, P. / Wang, M. / Standfuss, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6rqp.cif.gz | 106.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6rqp.ent.gz | 81.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6rqp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rq/6rqpftp://data.pdbj.org/pub/pdb/validation_reports/rq/6rqp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6rnjC  6rphC  6rqoC  6g7hS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / BR / Bacterioopsin / BO Mass: 25061.615 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Halobacterium salinarum NRC-1 (Halophile) / References: UniProt: P02945 | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-RET / Retinal  Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O / Feature type: SUBJECT OF INVESTIGATION Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O / Feature type: SUBJECT OF INVESTIGATION | ||||
| #3: Chemical | ChemComp-LI1 / 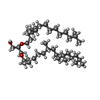  Mass: 639.130 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C42H86O3 Mass: 639.130 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C42H86O3#4: Chemical | 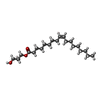  Mass: 356.540 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C21H40O4 Mass: 356.540 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C21H40O4#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.63 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: lipidic cubic phase / pH: 5.6 / Details: 100 mM Na/K Phosphate buffer pH 5.6 30 % PEG 2000 |
-Data collection
| Diffraction | Mean temperature: 298 K / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: May 6, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→30.8 Å / Num. obs: 22272 / % possible obs: 100 % / Redundancy: 1587.9 % / Biso Wilson estimate: 49.43 Å2 / CC1/2: 0.99 / R split: 0.026 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 1.8→1.85 Å / Redundancy: 198.2 % / Mean I/σ(I) obs: 1.17 / CC1/2: 0.31 / R split: 1.06 / % possible all: 100 |
| Serial crystallography sample delivery | Method: injection |
| Serial crystallography sample delivery injection | Carrier solvent: LCP / Jet diameter: 50 µm |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6G7H Resolution: 1.8→30.336 Å / SU ML: 0.06 / Cross valid method: THROUGHOUT / σ(F): 1.36 / Phase error: 21.07
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 184.55 Å2 / Biso mean: 73.7603 Å2 / Biso min: 45.71 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.8→30.336 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 10 / % reflection obs: 100 %
|
