 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9yd2: Ternary complex of DNA polymerase I from Bacillus stearothermophi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9yd2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Ternary complex of DNA polymerase I from Bacillus stearothermophilus, large fragment, bound to DNA containing a thymine dimer and dATP | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | REPLICATION / polymerase / complex | |||||||||
| Function / homology |  Function and homology information Function and homology information3'-5' exonuclease activity / DNA-templated DNA replication / double-strand break repair / DNA-directed DNA polymerase / DNA-directed DNA polymerase activity / nucleotide binding / DNA binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria)synthetic construct (others) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.249 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.249 Å | |||||||||
 Authors Authors | Wu, E.Y. / Walsh, A.R. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2026 Title: Structural basis for blockage of DNA synthesis by a thymine dimer lesion in a high-fidelity DNA polymerase Authors: Walsh, A.R. / Beese, L.S. / Wu, E.Y. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9yd2.cif.gz | 158.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9yd2.ent.gz | 113.8 KB | Display | PDB format |
| PDBx/mmJSON format | 9yd2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yd/9yd2ftp://data.pdbj.org/pub/pdb/validation_reports/yd/9yd2 | HTTPS FTP |
|---|
-Related structure data

-Links
 PDBj
PDBj








































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 66260.039 Da / Num. of mol.: 1 / Mutation: D598A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus stearothermophilus (bacteria)Gene: DPO1, polA / Production host: |
|---|
-DNA chain , 2 types, 2 molecules BC
| #2: DNA chain | Mass: 3391.250 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
|---|---|
| #3: DNA chain | Mass: 4824.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
-Non-polymers , 4 types, 103 molecules 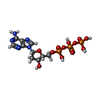



| #4: Chemical | ChemComp-DTP /  Mass: 491.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 491.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3 / Feature type: SUBJECT OF INVESTIGATION | ||
|---|---|---|---|
| #5: Chemical | ChemComp-MN /  Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn | ||
| #6: Chemical |  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.59 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop Details: 46% (v/v) saturated ammonium sulfate, 10 mM manganese sulfate (MnSO4), 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), 5% 2-methyl-1,3-propanediol (MPD), 1 mM 2',3'-dideoxyguanosine ...Details: 46% (v/v) saturated ammonium sulfate, 10 mM manganese sulfate (MnSO4), 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), 5% 2-methyl-1,3-propanediol (MPD), 1 mM 2',3'-dideoxyguanosine triphosphate, 5 mM 2'-deoxyadenosine triphosphate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-BM / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Mar 26, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.249→79.8 Å / Num. obs: 44763 / % possible obs: 99.3 % / Redundancy: 6.9 % / CC1/2: 0.999 / Rrim(I) all: 0.063 / Net I/σ(I): 21.64 |
| Reflection shell | Resolution: 2.249→2.31 Å / Redundancy: 4.6 % / Mean I/σ(I) obs: 1.42 / Num. unique obs: 3028 / CC1/2: 0.616 / Rrim(I) all: 1 / % possible all: 92.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.249→79.796 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.916 / SU B: 9.217 / SU ML: 0.215 / Cross valid method: FREE R-VALUE / ESU R: 0.258 / ESU R Free: 0.232 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 57.896 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.249→79.796 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
|
