 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9ul0: Wild-type Bacillus megaterium Penicillin G Acylase with Covalentl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9ul0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Wild-type Bacillus megaterium Penicillin G Acylase with Covalently Bound Phenylacetic Acid | ||||||
 Components Components | (Penicillin G ...) x 2 | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / N-terminal nucleophile hydrolase / Bacillus megaterium / Penicillin acylase / Covalently bound PAA | ||||||
| Function / homology |  Function and homology information Function and homology informationpenicillin amidase activity / penicillin amidase / antibiotic biosynthetic process / response to antibiotic / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  Priestia megaterium (bacteria) Priestia megaterium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.71 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.71 Å | ||||||
 Authors Authors | Kaewsasan, C. / Rojviriya, C. / Yuvaniyama, J. | ||||||
| Funding support |  Thailand, 1items Thailand, 1items
| ||||||
 Citation Citation | Journal: Acs Catalysis / Year: 2026 Title: Capturing Catalysis: Structural Insights into the Acyl-Enzyme Intermediate of Priestia megaterium Penicillin G Acylase Authors: Kaewsasan, C. / Rojviriya, C. / Oonanant, W. / Prathumrat, N. / Koinueng, W. / Yuvaniyama, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9ul0.cif.gz | 448.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9ul0.ent.gz | 299.4 KB | Display | PDB format |
| PDBx/mmJSON format | 9ul0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ul/9ul0ftp://data.pdbj.org/pub/pdb/validation_reports/ul/9ul0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9ub9C  9uewC  1pnmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
-Penicillin G ... , 2 types, 2 molecules AB
| #1: Protein | Mass: 24452.488 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: The protein is a heterodimer composed of two distinct subunits. The alpha subunit (chain A) encompasses residues G1 through S210, whereas the beta subunit (chain B) comprises residues S211 to K747. Source: (gene. exp.) Priestia megaterium (bacteria) / Gene: pac, pga / Plasmid: pBA402 / Production host: Priestia megaterium (bacteria) / Strain (production host): UN-cat / References: UniProt: Q60136, penicillin amidase |
|---|---|
| #2: Protein | Mass: 61475.883 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Priestia megaterium (bacteria) / Gene: pac, pga / Plasmid: pBA402 / Production host: Priestia megaterium (bacteria) / Strain (production host): UN-cat / References: UniProt: Q60136 |
-Non-polymers , 4 types, 1413 molecules 
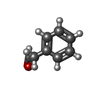


| #3: Chemical |  Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-HY1 / |  Mass: 120.149 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8O / Feature type: SUBJECT OF INVESTIGATION Mass: 120.149 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8O / Feature type: SUBJECT OF INVESTIGATION#5: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATION Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATION#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1407 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 43.9 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 1 mM Penicillin G, 0.150 M NaCl, 31% w/v PEG 4000, and 0.1 M imidazole (pH 6.5) PH range: 6.0-7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSRRC  / Beamline: BL13B1 / Wavelength: 0.99998 Å / Beamline: BL13B1 / Wavelength: 0.99998 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Aug 10, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.99998 Å / Relative weight: 1 |
| Reflection | Resolution: 1.71→30 Å / Num. obs: 80619 / % possible obs: 97.5 % / Redundancy: 3.5 % / Biso Wilson estimate: 15.95 Å2 / Rmerge(I) obs: 0.043 / Net I/σ(I): 23.45 |
| Reflection shell | Resolution: 1.71→1.77 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.183 / Mean I/σ(I) obs: 5.66 / Num. unique obs: 6551 / % possible all: 81.8 |
| Serial crystallography sample delivery | Method: fixed target |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1PNM Resolution: 1.71→28.67 Å / SU ML: 0.1228 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 15.0839 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.72 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.71→28.67 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 14.8542026283 Å / Origin y: 0.364144103219 Å / Origin z: 22.5958432987 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
