Entry Database : PDB / ID : 9sopTitle Tissue inhibitor of metalloproteinase-1 (TIMP-1) Metalloproteinase inhibitor 1 Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.95 Å Authors Shemy, A. / Voet, A. Funding support Organization Grant number Country Research Foundation - Flanders (FWO) 1S11123N Research Foundation - Flanders (FWO) 1S11125N
Journal : Acta Crystallogr D Struct Biol / Year : 2026Title : The human TIMP-1 unbound structure provides a platform for fragment screening.Authors : Shemy, A. / Van Broeckhoven, J. / Hellings, N. / Voet, A. History Deposition Sep 15, 2025 Deposition site / Processing site Revision 1.0 Sep 15, 2025 Provider / Type Revision 2.0 Apr 29, 2026 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Database references / Derived calculations / Non-polymer description / Polymer sequence / Refinement description / Source and taxonomy / Structure summary Category atom_site / chem_comp ... atom_site / chem_comp / chem_comp_atom / chem_comp_bond / citation / citation_author / entity / entity_poly / entity_poly_seq / entity_src_gen / pdbx_contact_author / pdbx_entity_nonpoly / pdbx_nonpoly_scheme / pdbx_poly_seq_scheme / pdbx_struct_assembly_gen / pdbx_struct_sheet_hbond / pdbx_unobs_or_zero_occ_residues / pdbx_validate_close_contact / pdbx_validate_peptide_omega / pdbx_validate_rmsd_angle / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_restr_ncs / refine_ls_shell / software / struct_asym / struct_conn / struct_mon_prot_cis / struct_ref / struct_ref_seq / struct_sheet / struct_sheet_order / struct_sheet_range Item _chem_comp.formula / _chem_comp.formula_weight ... _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.name / _chem_comp.pdbx_synonyms / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _entity.formula_weight / _entity.pdbx_description / _entity.pdbx_number_of_molecules / _entity_poly.pdbx_seq_one_letter_code / _entity_poly.pdbx_seq_one_letter_code_can / _entity_src_gen.pdbx_end_seq_num / _pdbx_entity_nonpoly.comp_id / _pdbx_entity_nonpoly.name / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_validate_rmsd_angle.angle_deviation / _pdbx_validate_rmsd_angle.angle_standard_deviation / _pdbx_validate_rmsd_angle.angle_target_value / _pdbx_validate_rmsd_angle.angle_value / _pdbx_validate_rmsd_angle.auth_atom_id_1 / _pdbx_validate_rmsd_angle.auth_atom_id_2 / _pdbx_validate_rmsd_angle.auth_atom_id_3 / _pdbx_validate_rmsd_angle.auth_comp_id_1 / _pdbx_validate_rmsd_angle.auth_comp_id_2 / _pdbx_validate_rmsd_angle.auth_comp_id_3 / _pdbx_validate_rmsd_angle.auth_seq_id_1 / _pdbx_validate_rmsd_angle.auth_seq_id_2 / _pdbx_validate_rmsd_angle.auth_seq_id_3 / _pdbx_validate_torsion.auth_asym_id / _pdbx_validate_torsion.auth_comp_id / _pdbx_validate_torsion.auth_seq_id / _pdbx_validate_torsion.phi / _pdbx_validate_torsion.psi / _refine.B_iso_mean / _refine.aniso_B[1][1] / _refine.aniso_B[1][3] / _refine.aniso_B[2][2] / _refine.aniso_B[3][3] / _refine.correlation_coeff_Fo_to_Fc / _refine.correlation_coeff_Fo_to_Fc_free / _refine.details / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_all / _refine.ls_wR_factor_R_free / _refine.ls_wR_factor_R_work / _refine.overall_SU_B / _refine.overall_SU_ML / _refine.pdbx_average_fsc_free / _refine.pdbx_average_fsc_work / _refine.pdbx_overall_ESU_R / _refine.pdbx_overall_ESU_R_Free / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_number_atoms_ligand / _refine_ls_restr_ncs.rms_dev_position / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_work / _refine_ls_shell.R_factor_all / _refine_ls_shell.pdbx_fsc_free / _refine_ls_shell.pdbx_fsc_work / _refine_ls_shell.wR_factor_R_work / _software.classification / _software.name / _software.version / _struct_conn.pdbx_dist_value / _struct_mon_prot_cis.pdbx_omega_angle / _struct_ref.pdbx_seq_one_letter_code / _struct_ref_seq.db_align_end / _struct_ref_seq.pdbx_auth_seq_align_end / _struct_ref_seq.seq_align_end Description Details: Some ligands around the biomolecule were re-modelled, particularly those with weak or discontinuous electron density or multiple alternative conformations. These sites are not relevant to ... Details Provider / Type
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Belgium, 2items
Belgium, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

 PDBj
PDBj








 Assembly
Assembly




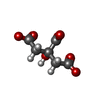
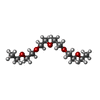


 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7
Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 250.332 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O5
Mass: 250.332 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O5 Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4
Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Sample preparation
Sample preparation / Beamline: I04-1 / Wavelength: 0.92205 Å
/ Beamline: I04-1 / Wavelength: 0.92205 Å Processing
Processing