 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9pqf: Xray crystal structure of Vientovirus FB Rep endonuclease domain ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9pqf | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Xray crystal structure of Vientovirus FB Rep endonuclease domain 2-114 with Mg2+ ion | |||||||||||||||||||||
 Components Components | Replication-associated protein | |||||||||||||||||||||
 Keywords Keywords | REPLICATION / Endonuclease / DNA binding protein / Viral protein | |||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationnucleotidyltransferase activity / endonuclease activity / DNA replication / RNA helicase activity / hydrolase activity / host cell nucleus / DNA binding / RNA binding / ATP binding / metal ion binding Similarity search - Function | |||||||||||||||||||||
| Biological species |  Human lung-associated vientovirus FB Human lung-associated vientovirus FB | |||||||||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | |||||||||||||||||||||
 Authors Authors | Montermoso, S. / Gupta, K. / Pumroy, R.A. / Moiseenkova-Bell, V. / Bushman, F.D. / Van Duyne, G.D. | |||||||||||||||||||||
| Funding support |  United States, 6items United States, 6items
| |||||||||||||||||||||
 Citation Citation | Journal: PLoS Pathog / Year: 2026 Title: Structures of nucleotide-bound Redondovirus Rep protein link conformation and function. Authors: Saira Montermoso / Kushol Gupta / Ruth Anne Pumroy / Vera Moiseenkova-Bell / Frederic D Bushman / Gregory D Van Duyne / Abstract: Circular Rep-encoding single-stranded DNA (CRESS-DNA) virus Rep proteins are multidomain enzymes that mediate viral DNA rolling-circle replication. Reps nick viral DNA to expose a 3' end for ...Circular Rep-encoding single-stranded DNA (CRESS-DNA) virus Rep proteins are multidomain enzymes that mediate viral DNA rolling-circle replication. Reps nick viral DNA to expose a 3' end for polymerase extension, provide an NTP-dependent helicase activity for DNA unwinding, and join nicked ends to form circular viral genomes. Here, we present the first structures of a Rep protein from the Redondoviridae family, a newly discovered family of human-associated CRESS-DNA viruses that replicates within the oral protozoan Entamoeba gingivalis. Using cryo-EM, we characterized the hexameric structures of a Redondovirus Rep helicase bound with ATPγS, representing the initial ATP-bound state, and with ADP, reflecting the protein state after hydrolysis. The ADP state, but not the ATP state of Rep shows a staircase arrangement of DNA-binding loops that plays a central role in current models for SF3 helicase function. Additionally, we determined a head-to-tail dodecameric structure of ATPγS-bound Rep, in which both the helicase and endonuclease domains are ordered. Conservation of residues involved in stabilizing the dodecamer suggest that this assembly may be functionally relevant for many CRESS-DNA viruses. The positioning of endonuclease domains in the Rep hexamer, combined with our biophysical analyses of Rep oligomerization, provide new insights into Rep function during viral replication. | |||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9pqf.cif.gz | 38.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9pqf.ent.gz | 24.4 KB | Display | PDB format |
| PDBx/mmJSON format | 9pqf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pq/9pqfftp://data.pdbj.org/pub/pdb/validation_reports/pq/9pqf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9pqjC  9pqmC  9pqoC  9pqqC  9pqrC  9pqtC C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 12903.830 Da / Num. of mol.: 1 / Mutation: T2S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human lung-associated vientovirus FB / Production host:  |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-MLI / 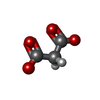  Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.02 Å3/Da / Density % sol: 39.2 % |
|---|---|
| Crystal grow | Temperature: 294.15 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: 100 mM ammonium malonate pH 5.8, 200 mM potassium fluoride, 15% w/v PEG 4000, 15% v/v ethylene glycol |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS-II / Beamline: 17-ID-1 / Wavelength: 0.9201 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Mar 27, 2024 / Details: KB bimorph mirrors |
| Radiation | Monochromator: Si(111) DCM / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9201 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→45.13 Å / Num. obs: 9971 / % possible obs: 100 % / Redundancy: 7.4 % / Biso Wilson estimate: 23.34 Å2 / CC1/2: 0.983 / CC star: 0.996 / Rmerge(I) obs: 0.194 / Rpim(I) all: 0.076 / Rrim(I) all: 0.209 / Net I/σ(I): 5.8 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 7.8 % / Rmerge(I) obs: 0.433 / Mean I/σ(I) obs: 1 / Num. unique obs: 475 / CC1/2: 0.816 / Rpim(I) all: 0.171 / Rrim(I) all: 0.466 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.8→45.13 Å / SU ML: 0.21 / Cross valid method: FREE R-VALUE / σ(F): 1.38 / Phase error: 19.9 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.91 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→45.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / % reflection obs: 100 %
|
