Software Name Version Classification dev_5660refinementdata scalingdata reductionphasingdata extraction
Refinement Method to determine structure / Resolution / SU ML / Cross valid method / σ(F) / Phase error / Stereochemistry target values Rfactor Num. reflection % reflection Rfree 0.2225 1710 4.97 % Rwork 0.1799 - - obs 0.1821 34400 97.8 %
Solvent computation Shrinkage radii / VDW probe radii / Solvent model Refinement step Cycle / Resolution Protein Nucleic acid Ligand Solvent Total Num. atoms 4900 0 64 148 5112
Refine LS restraints Refine-ID Type Dev ideal Number X-RAY DIFFRACTION f_bond_d0.003 5058 X-RAY DIFFRACTION f_angle_d0.566 6860 X-RAY DIFFRACTION f_dihedral_angle_d16.518 1832 X-RAY DIFFRACTION f_chiral_restr0.041 773 X-RAY DIFFRACTION f_plane_restr0.005 891
LS refinement shell Resolution (Å)Rfactor Rfree Num. reflection Rfree Rfactor Rwork Num. reflection Rwork Refine-ID % reflection obs (%)2.15-2.21 0.2988 131 0.234 2596 X-RAY DIFFRACTION 94 2.21-2.28 0.2485 159 0.2073 2675 X-RAY DIFFRACTION 97 2.28-2.37 0.2376 154 0.198 2710 X-RAY DIFFRACTION 98 2.37-2.46 0.2358 160 0.1949 2747 X-RAY DIFFRACTION 98 2.46-2.57 0.2379 123 0.1825 2726 X-RAY DIFFRACTION 98 2.57-2.71 0.2914 121 0.1857 2757 X-RAY DIFFRACTION 98 2.71-2.88 0.2446 145 0.1922 2742 X-RAY DIFFRACTION 99 2.88-3.1 0.2712 156 0.2092 2749 X-RAY DIFFRACTION 99 3.1-3.41 0.2328 141 0.185 2706 X-RAY DIFFRACTION 98 3.41-3.91 0.2036 136 0.1666 2770 X-RAY DIFFRACTION 99 3.91-4.92 0.1746 137 0.1461 2780 X-RAY DIFFRACTION 99 4.92-48.88 0.215 147 0.1875 2732 X-RAY DIFFRACTION 97
Refinement TLS params. Method / Refine-ID
Show large table (25 x 14) Hide large table ID L11 (°2 )L12 (°2 )L13 (°2 )L22 (°2 )L23 (°2 )L33 (°2 )S11 (Å °)S12 (Å °)S13 (Å °)S21 (Å °)S22 (Å °)S23 (Å °)S31 (Å °)S32 (Å °)S33 (Å °)T11 (Å2 )T12 (Å2 )T13 (Å2 )T22 (Å2 )T23 (Å2 )T33 (Å2 )Origin x (Å)Origin y (Å)Origin z (Å)1 1.3996 -0.9795 -0.7219 0.7282 0.24 1.9986 -0.0344 -0.4976 -0.1145 0.168 0.0174 0.1673 -0.0275 -0.1551 0.0272 0.3198 0.0258 -0.0123 0.464 -0.0268 0.3361 1.871 3.871 9.872 2 0.4767 -0.499 -0.1416 1.5459 0.3662 0.7897 0.0063 -0.1246 0.0226 0.2207 0.0231 0.0332 0.0319 0.0152 -0.0376 0.3079 0.025 -0.0063 0.3527 -0.0215 0.2496 8.046 -5.24 4.421 3 4.1332 -0.6243 -1.3971 0.7353 0.618 0.9822 -0.1057 -0.6228 -0.0797 0.2455 0.1637 -0.1443 0.0614 0.4369 -0.2104 0.393 0.048 -0.0847 0.4556 -0.0022 0.3875 8.271 0.124 16.253 4 2.9854 0.0222 -0.3794 2.726 -0.4656 3.0169 -0.1193 -0.0161 -0.255 0.1922 0.1908 -0.0457 0.0818 0.0537 -0.084 0.3503 0.0113 -0.0141 0.3487 0.0121 0.2929 -13.947 6.205 26.05 5 1.1286 -0.1371 -2.8156 0.3826 0.4386 6.9226 0.0525 0.1647 -0.0505 0.0356 -0.2684 0.1304 -0.7731 -0.7847 0.1223 0.3546 0.0884 -0.0167 0.4306 -0.0199 0.3524 -13.912 12.607 11.227 6 2.7644 -1.5832 -0.8174 1.873 0.8627 0.5671 -0.0435 -0.0677 0.3407 -0.1567 0.2018 -0.3888 -0.1354 0.0919 -0.1379 0.3381 -0.046 0.0269 0.3597 0.0034 0.3182 14.723 1.06 -7.701 7 2.003 -0.4773 -1.2859 2.5296 1.0325 1.1974 -0.2133 -0.1429 -0.045 0.2705 0.046 -0.3627 0.3659 -0.0671 0.1485 0.3237 -0.037 0.0224 0.2847 -0.0338 0.4224 22.414 -26.348 -23.616 8 2.1735 -0.382 -0.3876 1.2733 0.0792 1.947 0.0874 0.009 0.0085 -0.1354 0.0352 -0.1839 -0.0853 0.0506 -0.1397 0.2893 0.0253 0.0155 0.2658 -0.0068 0.319 23.689 -19.044 -20.868 9 1.5949 -0.5064 0.2418 0.3915 0.2682 2.0691 0.0252 0.0406 -0.3258 -0.0228 0.0701 0.0317 0.2327 0.0644 -0.0417 0.3227 0.0086 -0.0161 0.3107 0.0049 0.3821 9.806 -22.286 -8.85 10 3.0913 -2.2781 -1.0659 1.6757 0.6211 0.4704 -0.0687 -0.0751 -0.3878 -0.005 0.006 0.1194 0.0721 0.0582 0.0808 0.3393 -0.0359 -0.068 0.3265 -0.0043 0.498 9.938 -30.098 -16.23 11 1.6119 1.0676 0.026 4.5025 -0.8999 2.0431 -0.1397 -0.0206 -0.393 0.3971 0.0944 -1.1034 0.1682 -0.0519 0.0581 0.5134 0.0176 0.024 0.4679 -0.0766 0.667 36.261 -43.948 -25.92 12 0.1551 0.2754 -0.0836 3.826 0.4458 0.7852 -0.1039 0.4711 -0.1294 -1.2079 0.1786 -1.0004 0.0211 -0.0244 -0.1299 0.5409 -0.0722 0.2 0.5551 -0.1201 0.6079 37.706 -34.807 -33.332 13 2.3946 -0.8636 -1.0754 0.5501 0.219 1.2916 0.2329 0.3446 0.1277 -0.1304 -0.1263 -0.0284 -0.1867 -0.2067 -0.1755 0.355 0.0363 0.0235 0.2984 0.0382 0.345 11.909 -7.566 -18.878 14 1.4091 -0.579 0.76 2.6713 1.4065 2.4519 -0.0661 -0.4729 -0.4166 0.2473 0.4002 0.1953 0.1193 -0.2626 -0.2655 0.3607 0.034 -0.0411 0.8192 0.4965 1.2903 11.535 -15.193 -3.561
Refinement TLS group ID Refine-ID Refine TLS-ID Selection details Auth asym-ID Auth seq-ID 1 X-RAY DIFFRACTION 1 ( CHAIN A AND RESID 2:24 )A2 - 24 2 X-RAY DIFFRACTION 2 ( CHAIN A AND RESID 25:168 )A25 - 168 3 X-RAY DIFFRACTION 3 ( CHAIN A AND RESID 169:190 )A169 - 190 4 X-RAY DIFFRACTION 4 ( CHAIN A AND RESID 191:269 )A191 - 269 5 X-RAY DIFFRACTION 5 ( CHAIN A AND RESID 270:298 )A270 - 298 6 X-RAY DIFFRACTION 6 ( CHAIN A AND RESID 299:334 )A299 - 334 7 X-RAY DIFFRACTION 7 ( CHAIN B AND RESID 2:24 )B2 - 24 8 X-RAY DIFFRACTION 8 ( CHAIN B AND RESID 25:86 )B25 - 86 9 X-RAY DIFFRACTION 9 ( CHAIN B AND RESID 87:147 )B87 - 147 10 X-RAY DIFFRACTION 10 ( CHAIN B AND RESID 148:193 )B148 - 193 11 X-RAY DIFFRACTION 11 ( CHAIN B AND RESID 194:254 )B194 - 254 12 X-RAY DIFFRACTION 12 ( CHAIN B AND RESID 255:298 )B255 - 298 13 X-RAY DIFFRACTION 13 ( CHAIN B AND RESID 299:334 )B299 - 334 14 X-RAY DIFFRACTION 14 ( CHAIN A AND RESID 401:401 ) OR ( CHAIN B AND RESID 401
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Klebsiella aerogenes KCTC 2190 (bacteria)
Klebsiella aerogenes KCTC 2190 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 2items
United States, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads PDBj
PDBj

 Assembly
Assembly
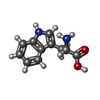




 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O2 / Feature type: SUBJECT OF INVESTIGATION
Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 194.226 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C8H18O5 / Comment: precipitant*YM
Mass: 194.226 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C8H18O5 / Comment: precipitant*YM Sample preparation
Sample preparation Processing
Processing