登録情報 データベース : PDB / ID : 9mrwタイトル Functional Implications of Hexameric Dynamics in SARS-CoV-2 Nsp15 Uridylate-specific endoribonuclease nsp15 キーワード / / / / / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / 生物種 手法 / / / 解像度 : 3 Å データ登録者 Ketawala, G.K. / Sonowal, M. / Schrag, L. / Fromme, R. / Botha, S. / Fromme, P. 資金援助 組織 認可番号 国 National Science Foundation (NSF, United States) STC-1231306 National Science Foundation (NSF, United States) 2031343
ジャーナル : Protein Sci. / 年 : 2025タイトル : Functional implications of hexameric dynamics in SARS-CoV-2 Nsp15.著者: Sonowal, M. / Ketawala, G. / Nagaratnam, N. / Logeswaran, D. / Basu, S. / de Sanctis, D. / Orlans, J. / Rose, S.L. / Jernigan, R.J. / Hu, H. / Aguilar, J.D.M. / Ranaweera, M.E. / Zacks, M.A. ... 著者 : Sonowal, M. / Ketawala, G. / Nagaratnam, N. / Logeswaran, D. / Basu, S. / de Sanctis, D. / Orlans, J. / Rose, S.L. / Jernigan, R.J. / Hu, H. / Aguilar, J.D.M. / Ranaweera, M.E. / Zacks, M.A. / Chen, J.J. / Hansen, D.T. / Schrag, L.G. / Fromme, R. / Botha, S. / Fromme, P. 履歴 登録 2025年1月8日 登録サイト / 処理サイト 改定 1.0 2025年6月4日 Provider / タイプ
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Severe acute respiratory syndrome coronavirus 2 (ウイルス)
Severe acute respiratory syndrome coronavirus 2 (ウイルス) X線回折 /
X線回折 /  データ登録者
データ登録者 米国, 2件
米国, 2件  引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード

 PDBj
PDBj







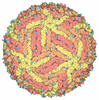
 集合体
集合体


 試料調製
試料調製 / ビームライン: ID29 / 波長: 1.075 Å
/ ビームライン: ID29 / 波長: 1.075 Å 解析
解析