 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9hd0: Crystal structure of human TRF1 TRFH domain in complex with compo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9hd0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human TRF1 TRFH domain in complex with compound 55 | ||||||
 Components Components | Telomeric repeat-binding factor 1 | ||||||
 Keywords Keywords | PROTEIN BINDING / Telomere / Shelterin / Inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of shelterin complex assembly / negative regulation of establishment of protein localization to telomere / negative regulation of establishment of RNA localization to telomere / negative regulation of establishment of protein-containing complex localization to telomere / negative regulation of telomere maintenance via semi-conservative replication / negative regulation of telomeric D-loop disassembly / meiotic telomere clustering / telomeric D-loop disassembly / shelterin complex / Telomere C-strand synthesis initiation ...positive regulation of shelterin complex assembly / negative regulation of establishment of protein localization to telomere / negative regulation of establishment of RNA localization to telomere / negative regulation of establishment of protein-containing complex localization to telomere / negative regulation of telomere maintenance via semi-conservative replication / negative regulation of telomeric D-loop disassembly / meiotic telomere clustering / telomeric D-loop disassembly / shelterin complex / Telomere C-strand synthesis initiation / double-stranded telomeric DNA binding / t-circle formation / ankyrin repeat binding / Telomere C-strand (Lagging Strand) Synthesis / G-rich strand telomeric DNA binding / nuclear telomere cap complex / telomere capping / Polymerase switching on the C-strand of the telomere / Processive synthesis on the C-strand of the telomere / Removal of the Flap Intermediate from the C-strand / DNA binding, bending / negative regulation of telomere maintenance via telomere lengthening / negative regulation of DNA replication / telomeric repeat DNA binding / negative regulation of telomere maintenance via telomerase / positive regulation of telomere maintenance / Telomere Extension By Telomerase / telomere maintenance via telomerase / Packaging Of Telomere Ends / Recognition and association of DNA glycosylase with site containing an affected purine / Cleavage of the damaged purine / Recognition and association of DNA glycosylase with site containing an affected pyrimidine / Cleavage of the damaged pyrimidine / telomere maintenance / Inhibition of DNA recombination at telomere / Meiotic synapsis / DNA Damage/Telomere Stress Induced Senescence / spindle / fibrillar center / microtubule binding / chromosome, telomeric region / nuclear body / response to xenobiotic stimulus / cell division / nucleolus / protein homodimerization activity / DNA binding / nucleoplasm / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å | ||||||
 Authors Authors | Casale, G. / Le Bihan, Y.-V. / van Montfort, R.L.M. / Guettler, S. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Sci Rep / Year: 2025 Title: Discovery of first-in-class inhibitors of the TRF1:TIN2 protein:protein interaction by fragment screening. Authors: Casale, G. / Liu, M. / Le Bihan, Y.V. / Inian, O. / Stammers, E. / Caldwell, J. / van Montfort, R.L.M. / Collins, I. / Guettler, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9hd0.cif.gz | 98.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9hd0.ent.gz | 73.6 KB | Display | PDB format |
| PDBx/mmJSON format | 9hd0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hd/9hd0ftp://data.pdbj.org/pub/pdb/validation_reports/hd/9hd0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9hclC  9hcmC  9hcnC  9hcpC  9hcqC  9hcrC  9hcsC  9hctC  9hcuC  9hcvC  9hcwC  9hcxC  9hcyC  9hczC  9hd1C  9hd2C  9hd3C  9hd9C  9hdaC  9hf4C  9hf6C  9hf7C  9hf8C  9hf9C  9hfaC  9hfbC  9hfcC  9hfdC  9hfeC  9hffC  9hfgC  9hfhC  9hfiC  9hhkC  9hlpC  9hlqC  9hlrC  9hltC  9hluC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25556.039 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TERF1, PIN2, TRBF1, TRF, TRF1 / Plasmid: pET His6 MBP Asn10 TEV LIC / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-A9E / 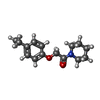  Mass: 247.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21NO2 / Feature type: SUBJECT OF INVESTIGATION Mass: 247.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21NO2 / Feature type: SUBJECT OF INVESTIGATION | ||||||||
| #3: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 126 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 126 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.94 Å3/Da / Density % sol: 36.64 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 6 Details: ...Details: SNAQVQVGAPEEEEEEEEDAGLVAEAEAVAAGWMLDFLCLSLCRAFRDGRSEDFRRTRNSAEAIIHGLSSLTACQLRTIYICQFLTRIAAGKTLDAQFENDERITPLESALMIWGSIEKEHDKLHEEIQNLIKIQAIAVCMENGNFKEAEEVFERIFGDPNSHMPFKSKLLMIISQKDTFHSFFQHFSYNHMMEKIKSYVNYVLSEKSSTFLMKAAAKVVESKR |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04-1 / Wavelength: 0.9212 Å |
| Detector | Type: DECTRIS EIGER2 XE 9M / Detector: PIXEL / Date: Jul 6, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9212 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→49.08 Å / Num. obs: 15989 / % possible obs: 100 % / Redundancy: 13.7 % / CC1/2: 0.989 / Rmerge(I) obs: 0.142 / Rpim(I) all: 0.04 / Rrim(I) all: 0.148 / Χ2: 0.82 / Net I/σ(I): 9.5 / Num. measured all: 218993 |
| Reflection shell | Resolution: 1.93→1.98 Å / % possible obs: 100 % / Redundancy: 12.6 % / Rmerge(I) obs: 2.409 / Num. measured all: 12842 / Num. unique obs: 1021 / CC1/2: 0.685 / Rpim(I) all: 0.703 / Rrim(I) all: 2.512 / Χ2: 0.76 / Net I/σ(I) obs: 1.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.93→48.97 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.924 / SU R Cruickshank DPI: 0.174 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.192 / SU Rfree Blow DPI: 0.15 / SU Rfree Cruickshank DPI: 0.143
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.28 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.24 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.93→48.97 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.93→1.95 Å / Total num. of bins used: 40
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 3.6591 Å / Origin y: 19.4817 Å / Origin z: 16.0599 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: {A|63 - 266} |
