 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9hci: structure of the double Cys-substituted cross-linked AcrB variant... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9hci | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | structure of the double Cys-substituted cross-linked AcrB variant S562C_T837C | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | TRANSPORT PROTEIN / RND multidrug efflux pump / antimicrobial resistance / intermediate conformational state / crosslinked structure | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationalkane transmembrane transporter activity / alkane transport / enterobactin transport / enterobactin transmembrane transporter activity / xenobiotic detoxification by transmembrane export across the cell outer membrane / periplasmic side of plasma membrane / efflux pump complex / Iron assimilation using enterobactin / bile acid transmembrane transporter activity / Antimicrobial resistance ...alkane transmembrane transporter activity / alkane transport / enterobactin transport / enterobactin transmembrane transporter activity / xenobiotic detoxification by transmembrane export across the cell outer membrane / periplasmic side of plasma membrane / efflux pump complex / Iron assimilation using enterobactin / bile acid transmembrane transporter activity / Antimicrobial resistance / bile acid and bile salt transport / Secretion of toxins / efflux transmembrane transporter activity / xenobiotic transmembrane transporter activity / fatty acid transport / xenobiotic transport / response to toxic substance / response to xenobiotic stimulus / response to antibiotic / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||||||||
| Biological species |  synthetic construct (others) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||||||||
 Authors Authors | Brandstaetter, L. / Mueller, R.T. / Pos, K.M. | ||||||||||||
| Funding support |  Switzerland, Switzerland,  Germany, 3items Germany, 3items
| ||||||||||||
 Citation Citation | Journal: NPJ Antimicrob Resist / Year: 2026 Title: Molecular mechanism of transition-state inhibitors of bacterial antibiotic efflux pumps. Authors: Bornsen, C. / Muller, R.T. / Vieira Da Cruz, A. / Jimenez-Castellanos, J.C. / Meurillon, V. / Brandstatter, L. / Lodinsky, E.V. / Athar, M. / Vargiu, A.V. / Hartkoorn, R.C. / Flipo, M. / ...Authors: Bornsen, C. / Muller, R.T. / Vieira Da Cruz, A. / Jimenez-Castellanos, J.C. / Meurillon, V. / Brandstatter, L. / Lodinsky, E.V. / Athar, M. / Vargiu, A.V. / Hartkoorn, R.C. / Flipo, M. / Frangakis, A.S. / Pos, K.M. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9hci.cif.gz | 1.3 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9hci.ent.gz | 1.1 MB | Display | PDB format |
| PDBx/mmJSON format | 9hci.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hc/9hciftp://data.pdbj.org/pub/pdb/validation_reports/hc/9hci | HTTPS FTP |
|---|
-Related structure data

-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 5 molecules ABCDE
| #1: Protein | Mass: 114754.398 Da / Num. of mol.: 3 / Mutation: S562C, T837C Source method: isolated from a genetically manipulated source Details: disulfide bond between S562C and T837C / Source: (gene. exp.) #2: Protein | Mass: 18317.566 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Plasmid: pQE30 / Details (production host): pQE30_110819 / Production host: |
|---|
-Sugars , 1 types, 8 molecules 
| #3: Sugar | ChemComp-LMT /  Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM |
|---|
-Non-polymers , 9 types, 1252 molecules 


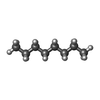
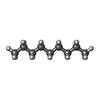


| #4: Chemical | ChemComp-D10 /  Mass: 142.282 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C10H22 Mass: 142.282 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C10H22#5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-D12 /  Mass: 170.335 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C12H26 Mass: 170.335 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C12H26#7: Chemical |  Mass: 86.175 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14 Mass: 86.175 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14#8: Chemical | ChemComp-C14 / |  Mass: 198.388 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30 Mass: 198.388 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30#9: Chemical |  Mass: 114.229 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18 Mass: 114.229 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18#10: Chemical | ChemComp-DD9 / |  Mass: 128.255 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H20 Mass: 128.255 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H20#11: Chemical | ChemComp-SO4 / |  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#12: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1215 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | N |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.87 Å3/Da / Density % sol: 68.23 % / Description: Rod-shaped crystals |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.05M ADA, pH 6.5, 0.2M ammonium sulfate, 8% PEG4000, 5.1% Glycerol |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS / Beamline: X06SA / Wavelength: 0.99997 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 19, 2007 |
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.99997 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→47.69 Å / Num. obs: 181397 / % possible obs: 99.94 % / Redundancy: 8.4 % / CC1/2: 0.982 / Net I/σ(I): 7.09 |
| Reflection shell | Resolution: 2.6→2.693 Å / Num. unique obs: 17959 / CC1/2: 0.683 / % possible all: 99.99 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.6→47.69 Å / Cor.coef. Fo:Fc: 0.931 / Cor.coef. Fo:Fc free: 0.904 / SU B: 18.063 / SU ML: 0.196 / Cross valid method: THROUGHOUT / ESU R: 0.339 / ESU R Free: 0.244 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: U VALUES : WITH TLS ADDED HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES : RESIDUAL ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.1 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 45.783 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→47.69 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.667 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
