 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 9giu | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Cryo-EM structure of human SLC45A4 in detergent | |||||||||||||||||||||||||||
 要素 要素 | Solute carrier family 45 member 4 | |||||||||||||||||||||||||||
 キーワード キーワード | TRANSPORT PROTEIN / SLC / MFS / Polyamine / Spermidine | |||||||||||||||||||||||||||
| 機能・相同性 | sucrose:proton symporter activity / sucrose transport / Major facilitator superfamily / Major Facilitator Superfamily / MFS transporter superfamily / membrane / 1,2-Distearoyl-sn-glycerophosphoethanolamine / CHOLESTEROL HEMISUCCINATE / Solute carrier family 45 member 4 機能・相同性情報 機能・相同性情報 | |||||||||||||||||||||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||||||||||||||||||||
| 手法 | 電子顕微鏡法 / 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.8 Å | |||||||||||||||||||||||||||
 データ登録者 データ登録者 | Markusson, S. / Deme, J.C. / Lea, S.M. / Newstead, S. | |||||||||||||||||||||||||||
| 資金援助 |  英国, 英国,  米国, 3件 米国, 3件
| |||||||||||||||||||||||||||
 引用 引用 | ジャーナル: To Be Published タイトル: Cryo-EM structure of human SLC45A4 in detergent 著者: Markusson, S. / Deme, J.C. / Lea, S.M. / Newstead, S. #1: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2018 タイトル: Real-space refinement in PHENIX for cryo-EM and crystallography. 著者: Pavel V Afonine / Billy K Poon / Randy J Read / Oleg V Sobolev / Thomas C Terwilliger / Alexandre Urzhumtsev / Paul D Adams /  要旨: This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast ...This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps. #2: ジャーナル: Acta Crystallogr D Biol Crystallogr / 年: 2010 タイトル: Features and development of Coot. 著者: P Emsley / B Lohkamp / W G Scott / K Cowtan / 要旨: Coot is a molecular-graphics application for model building and validation of biological macromolecules. The program displays electron-density maps and atomic models and allows model manipulations ...Coot is a molecular-graphics application for model building and validation of biological macromolecules. The program displays electron-density maps and atomic models and allows model manipulations such as idealization, real-space refinement, manual rotation/translation, rigid-body fitting, ligand search, solvation, mutations, rotamers and Ramachandran idealization. Furthermore, tools are provided for model validation as well as interfaces to external programs for refinement, validation and graphics. The software is designed to be easy to learn for novice users, which is achieved by ensuring that tools for common tasks are 'discoverable' through familiar user-interface elements (menus and toolbars) or by intuitive behaviour (mouse controls). Recent developments have focused on providing tools for expert users, with customisable key bindings, extensions and an extensive scripting interface. The software is under rapid development, but has already achieved very widespread use within the crystallographic community. The current state of the software is presented, with a description of the facilities available and of some of the underlying methods employed. #3: ジャーナル: Nat Methods / 年: 2017 タイトル: cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. 著者: Ali Punjani / John L Rubinstein / David J Fleet / Marcus A Brubaker /  要旨: Single-particle electron cryomicroscopy (cryo-EM) is a powerful method for determining the structures of biological macromolecules. With automated microscopes, cryo-EM data can often be obtained in a ...Single-particle electron cryomicroscopy (cryo-EM) is a powerful method for determining the structures of biological macromolecules. With automated microscopes, cryo-EM data can often be obtained in a few days. However, processing cryo-EM image data to reveal heterogeneity in the protein structure and to refine 3D maps to high resolution frequently becomes a severe bottleneck, requiring expert intervention, prior structural knowledge, and weeks of calculations on expensive computer clusters. Here we show that stochastic gradient descent (SGD) and branch-and-bound maximum likelihood optimization algorithms permit the major steps in cryo-EM structure determination to be performed in hours or minutes on an inexpensive desktop computer. Furthermore, SGD with Bayesian marginalization allows ab initio 3D classification, enabling automated analysis and discovery of unexpected structures without bias from a reference map. These algorithms are combined in a user-friendly computer program named cryoSPARC (http://www.cryosparc.com). #4: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2018タイトル: ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. 著者: Tristan Ian Croll / 要旨: This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density ...This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density maps. ISOLDE combines interactive molecular-dynamics flexible fitting with modern molecular-graphics visualization and established structural biology libraries to provide an immersive interface wherein the model constantly acts to maintain physically realistic conformations as the user interacts with it by directly tugging atoms with a mouse or haptic interface or applying/removing restraints. In addition, common validation tasks are accelerated and visualized in real time. Using the recently described 3.8 Å resolution cryo-EM structure of the eukaryotic minichromosome maintenance (MCM) helicase complex as a case study, it is demonstrated how ISOLDE can be used alongside other modern refinement tools to avoid common pitfalls of low-resolution modelling and improve the quality of the final model. A detailed analysis of changes between the initial and final model provides a somewhat sobering insight into the dangers of relying on a small number of validation metrics to judge the quality of a low-resolution model. #5: ジャーナル: Elife / 年: 2018タイトル: New tools for automated high-resolution cryo-EM structure determination in RELION-3. 著者: Jasenko Zivanov / Takanori Nakane / Björn O Forsberg / Dari Kimanius / Wim Jh Hagen / Erik Lindahl / Sjors Hw Scheres /   要旨: Here, we describe the third major release of RELION. CPU-based vector acceleration has been added in addition to GPU support, which provides flexibility in use of resources and avoids memory ...Here, we describe the third major release of RELION. CPU-based vector acceleration has been added in addition to GPU support, which provides flexibility in use of resources and avoids memory limitations. Reference-free autopicking with Laplacian-of-Gaussian filtering and execution of jobs from python allows non-interactive processing during acquisition, including 2D-classification, model generation and 3D-classification. Per-particle refinement of CTF parameters and correction of estimated beam tilt provides higher resolution reconstructions when particles are at different heights in the ice, and/or coma-free alignment has not been optimal. Ewald sphere curvature correction improves resolution for large particles. We illustrate these developments with publicly available data sets: together with a Bayesian approach to beam-induced motion correction it leads to resolution improvements of 0.2-0.7 Å compared to previous RELION versions. #6: ジャーナル: J Struct Biol / 年: 2020 タイトル: WITHDRAWN: SIMPLE 3.0. Stream single-particle cryo-EM analysis in real time. 著者: Joseph Caesar / Cyril F Reboul / Chiara Machello / Simon Kiesewetter / Molly L Tang / Justin C Deme / Steven Johnson / Dominika Elmlund / Susan M Lea / Hans Elmlund /  | |||||||||||||||||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 9giu.cif.gz | 122.2 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb9giu.ent.gz | 88.7 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 9giu.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 9giu_validation.pdf.gz | 1.5 MB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 9giu_full_validation.pdf.gz | 1.5 MB | 表示 | |
| XML形式データ | 9giu_validation.xml.gz | 37.4 KB | 表示 | |
| CIF形式データ | 9giu_validation.cif.gz | 52.5 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/gi/9giuftp://data.pdbj.org/pub/pdb/validation_reports/gi/9giu | HTTPS FTP |
-関連構造データ

-リンク
 PDBj
PDBj


- 集合体
集合体
| 登録構造単位 | 
|
|---|---|
| 1 |
|
-要素
| #1: タンパク質 | 分子量: 84750.562 Da / 分子数: 1 / 由来タイプ: 組換発現 / 由来: (組換発現) Homo sapiens (ヒト) / 遺伝子: SLC45A4, KIAA1126 / 発現宿主:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: 化合物 | ChemComp-Y01 / 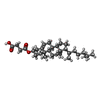  分子量: 486.726 Da / 分子数: 7 / 由来タイプ: 合成 / 式: C31H50O4 分子量: 486.726 Da / 分子数: 7 / 由来タイプ: 合成 / 式: C31H50O4#3: 化合物 | ChemComp-3PE / |   分子量: 748.065 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C41H82NO8P / コメント: リン脂質*YM 分子量: 748.065 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C41H82NO8P / コメント: リン脂質*YM#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 17 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 17 / 由来タイプ: 天然 / 式: H2O研究の焦点であるリガンドがあるか | N | Has protein modification | N | |
-実験情報
-実験
| 実験 | 手法: 電子顕微鏡法 |
|---|---|
| EM実験 | 試料の集合状態: PARTICLE / 3次元再構成法: 単粒子再構成法 |
- 試料調製
試料調製
| 構成要素 | 名称: SLC45A4 / タイプ: COMPLEX / Entity ID: #1 / 由来: RECOMBINANT |
|---|---|
| 分子量 | 実験値: NO |
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
| 由来(組換発現) | 生物種: |
| 緩衝液 | pH: 7.5 |
| 試料 | 濃度: 0.3 mg/ml / 包埋: NO / シャドウイング: NO / 染色: NO / 凍結: YES |
| 試料支持 | グリッドの材料: GOLD / グリッドのサイズ: 300 divisions/in. / グリッドのタイプ: Quantifoil R1.2/1.3 |
| 急速凍結 | 装置: FEI VITROBOT MARK IV / 凍結剤: ETHANE / 湿度: 100 % / 凍結前の試料温度: 283 K |
- 電子顕微鏡撮影
電子顕微鏡撮影
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
|---|---|
| 顕微鏡 | モデル: FEI TITAN KRIOS |
| 電子銃 | 電子線源:  FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM |
| 電子レンズ | モード: BRIGHT FIELD / 倍率(公称値): 165000 X / 最大 デフォーカス(公称値): 2500 nm / 最小 デフォーカス(公称値): 600 nm |
| 撮影 | 電子線照射量: 57.6 e/Å2 フィルム・検出器のモデル: FEI FALCON IV (4k x 4k) 実像数: 21269 |
| 電子光学装置 | エネルギーフィルター名称: TFS Selectris X / エネルギーフィルタースリット幅: 10 eV |
- 解析
解析
| CTF補正 | タイプ: PHASE FLIPPING AND AMPLITUDE CORRECTION |
|---|---|
| 対称性 | 点対称性: C1 (非対称) |
| 3次元再構成 | 解像度: 2.8 Å / 解像度の算出法: FSC 0.143 CUT-OFF / 粒子像の数: 700436 / 対称性のタイプ: POINT |
| 精密化 | 交差検証法: NONE |
