- PDB-9bbh: Co-crystal structure of human DDB1 bound to fragment UB028670 -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 9bbh
Title
Co-crystal structure of human DDB1 bound to fragment UB028670
Components
DNA damage-binding protein 1
Keywords
LIGASE / WD-repeat / WDR / DDB1 / SGC
Function / homology
Function and homology information
positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / Cul4A-RING E3 ubiquitin ligase complex / Cul4-RING E3 ubiquitin ligase complex / Cul4B-RING E3 ubiquitin ligase complex ...positive regulation by virus of viral protein levels in host cell / spindle assembly involved in female meiosis / epigenetic programming in the zygotic pronuclei / UV-damage excision repair / biological process involved in interaction with symbiont / regulation of mitotic cell cycle phase transition / WD40-repeat domain binding / Cul4A-RING E3 ubiquitin ligase complex / Cul4-RING E3 ubiquitin ligase complex / Cul4B-RING E3 ubiquitin ligase complex / ubiquitin ligase complex scaffold activity / negative regulation of reproductive process / negative regulation of developmental process / viral release from host cell / cullin family protein binding / ectopic germ cell programmed cell death / positive regulation of viral genome replication / proteasomal protein catabolic process / positive regulation of gluconeogenesis / nucleotide-excision repair / sperm end piece / regulation of circadian rhythm / Recognition of DNA damage by PCNA-containing replication complex / DNA Damage Recognition in GG-NER / Dual Incision in GG-NER / Wnt signaling pathway / Transcription-Coupled Nucleotide Excision Repair (TC-NER) / Formation of TC-NER Pre-Incision Complex / Formation of Incision Complex in GG-NER / Dual incision in TC-NER / positive regulation of protein catabolic process / Gap-filling DNA repair synthesis and ligation in TC-NER / cellular response to UV / rhythmic process / site of double-strand break / sperm principal piece / Neddylation / sperm midpiece / ubiquitin-dependent protein catabolic process / damaged DNA binding / proteasome-mediated ubiquitin-dependent protein catabolic process / protein-macromolecule adaptor activity / chromosome, telomeric region / protein ubiquitination / DNA repair / apoptotic process / DNA damage response / negative regulation of apoptotic process / protein-containing complex binding / nucleolus / protein-containing complex / : / DNA binding / extracellular exosome / nucleoplasm / nucleus / cytoplasm Similarity search - Function
: / RSE1/DDB1/CPSF1 second beta-propeller / Cleavage/polyadenylation specificity factor, A subunit, C-terminal / Cleavage/polyadenylation specificity factor, A subunit, N-terminal / : / CPSF A subunit region / RSE1/DDB1/CPSF1 first beta-propeller / WD40-repeat-containing domain superfamily / WD40/YVTN repeat-like-containing domain superfamily Similarity search - Domain/homology
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads PDBj
PDBj



 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm) / References: UniProt: Q16531
Spodoptera frugiperda (fall armyworm) / References: UniProt: Q16531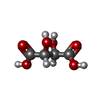




 Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6
Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 147.174 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C9H9NO / Feature type: SUBJECT OF INVESTIGATION
Mass: 147.174 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C9H9NO / Feature type: SUBJECT OF INVESTIGATION Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Num. of mol.: 5 / Source method: obtained synthetically
Num. of mol.: 5 / Source method: obtained synthetically Sample preparation
Sample preparation Processing
Processing