| Entry | Database: PDB / ID: 8r5d
|
|---|
| Title | Crystal structure of human TRIM7 PRYSPRY domain |
|---|
 Components Components | E3 ubiquitin-protein ligase TRIM7 |
|---|
 Keywords Keywords | LIGASE / TRIM7 / E3-Ligase / Structural Genomics / Structural Genomics Consortium / SGC |
|---|
| Function / homology |  Function and homology information Function and homology information
antiviral innate immune response / RING-type E3 ubiquitin transferase / ubiquitin protein ligase activity / protein ubiquitination / innate immune response / Golgi apparatus / zinc ion binding / nucleus / cytoplasmSimilarity search - Function zinc finger of C3HC4-type, RING / Zinc finger, B-box, chordata / : / SPRY-associated domain / SPRY-associated / PRY / B-box zinc finger / Butyrophylin-like, SPRY domain / B-Box-type zinc finger / B-box-type zinc finger ...zinc finger of C3HC4-type, RING / Zinc finger, B-box, chordata / : / SPRY-associated domain / SPRY-associated / PRY / B-box zinc finger / Butyrophylin-like, SPRY domain / B-Box-type zinc finger / B-box-type zinc finger / Zinc finger B-box type profile. / SPRY domain / B30.2/SPRY domain / B30.2/SPRY domain profile. / B30.2/SPRY domain superfamily / Domain in SPla and the RYanodine Receptor. / SPRY domain / Zinc finger, RING-type, conserved site / Zinc finger RING-type signature. / Ring finger / Zinc finger RING-type profile. / Zinc finger, RING-type / Concanavalin A-like lectin/glucanase domain superfamily / Zinc finger, RING/FYVE/PHD-typeSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å |
|---|
 Authors Authors | Munoz Sosa, C.J. / Kraemer, A. / Knapp, S. / Structural Genomics Consortium (SGC) |
|---|
| Funding support |  Switzerland, 1items Switzerland, 1items | Organization | Grant number | Country |
|---|
| Innovative Medicines Initiative | 875510 | Switzerland |
|
|---|
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2024
Title: A C-Degron Structure-Based Approach for the Development of Ligands Targeting the E3 Ligase TRIM7.
Authors: Munoz Sosa, C.J. / Lenz, C. / Hamann, A. / Farges, F. / Dopfer, J. / Kramer, A. / Cherkashyna, V. / Tarnovskiy, A. / Moroz, Y.S. / Proschak, E. / Nemec, V. / Muller, S. / Saxena, K. / Knapp, S. |
|---|
| History | | Deposition | Nov 16, 2023 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jan 17, 2024 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 31, 2024 | Group: Database references / Category: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
 PDBj
PDBj


 Assembly
Assembly

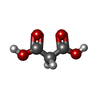
 Mass: 104.061 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H4O4
Mass: 104.061 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H4O4
 Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 216 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 216 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing