 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8r2p: YZwIdeal x16 a scaffold for cryo-EM of small proteins of interest... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8r2p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | YZwIdeal x16 a scaffold for cryo-EM of small proteins of interest crystallizing in space group 19 (P 21 21 21) | |||||||||
 Components Components | Putrescine aminotransferase,Immunoglobulin G-binding protein A | |||||||||
 Keywords Keywords | STRUCTURAL PROTEIN / Scaffold Fusion protein | |||||||||
| Function / homology |  Function and homology information Function and homology informationdiamine transaminase / putrescine-2-oxoglutarate transaminase / diamine transaminase activity / putrescine--2-oxoglutarate transaminase activity / L-lysine catabolic process / putrescine catabolic process / IgG binding / pyridoxal phosphate binding / protein homodimerization activity / extracellular region ...diamine transaminase / putrescine-2-oxoglutarate transaminase / diamine transaminase activity / putrescine--2-oxoglutarate transaminase activity / L-lysine catabolic process / putrescine catabolic process / IgG binding / pyridoxal phosphate binding / protein homodimerization activity / extracellular region / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.22 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.22 Å | |||||||||
 Authors Authors | Moche, M. / Friberg, O. / Nygren, P.A. / Nilvebrant, J. | |||||||||
| Funding support |  Sweden, 2items Sweden, 2items
| |||||||||
 Citation Citation | Journal: To Be Published Title: Engineered imaging scaffolds for cryo-EM of small proteins of interest. Authors: Moche, M. / Nygren, P.A. / Nilvebrant, J. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8r2p.cif.gz | 408.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8r2p.ent.gz | 328.6 KB | Display | PDB format |
| PDBx/mmJSON format | 8r2p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 8r2p_validation.pdf.gz | 488.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 8r2p_full_validation.pdf.gz | 514.6 KB | Display | |
| Data in XML | 8r2p_validation.xml.gz | 96.1 KB | Display | |
| Data in CIF | 8r2p_validation.cif.gz | 131.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r2/8r2pftp://data.pdbj.org/pub/pdb/validation_reports/r2/8r2p | HTTPS FTP |
-Related structure data

-Links
 PDBj
PDBj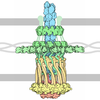
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
| #1: Protein | Mass: 57054.121 Da / Num. of mol.: 4 / Mutation: G487A Source method: isolated from a genetically manipulated source Details: The polypeptide chain consists of P42588_7-457 and P38507_160-269 connected by a linker. P38507 is mutated at Gly240Ala corresponding to residue 487 in the model.,The polypeptide chain ...Details: The polypeptide chain consists of P42588_7-457 and P38507_160-269 connected by a linker. P38507 is mutated at Gly240Ala corresponding to residue 487 in the model.,The polypeptide chain consists of P42588_7-457 and P38507_160-269 connected by a linker. P38507 is mutated at Gly240Ala corresponding to residue 487 in the model. Source: (gene. exp.) References: UniProt: P42588, UniProt: P38507, putrescine-2-oxoglutarate transaminase, diamine transaminase #2: Chemical | ChemComp-PLP /   Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1507 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1507 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.29 Å3/Da / Density % sol: 62.56 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 50% w/v PEG200, 0.2M MgCl2 and 0.1M Sodium Cacodylate buffer pH 6.5 PH range: 6.5-7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX IV / Beamline: BioMAX / Wavelength: 0.976246 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Sep 17, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976246 Å / Relative weight: 1 |
| Reflection | Resolution: 2.217→48.91 Å / Num. obs: 61481 / % possible obs: 41.1 % / Redundancy: 6.6 % / CC1/2: 0.997 / Rmerge(I) obs: 0.167 / Rpim(I) all: 0.07 / Rrim(I) all: 0.182 / Net I/σ(I): 8.1 |
| Reflection shell | Resolution: 2.217→2.425 Å / Redundancy: 5.12 % / Rmerge(I) obs: 1.041 / Mean I/σ(I) obs: 1.68 / Num. unique obs: 3073 / CC1/2: 0.665 / Rpim(I) all: 0.49 / Rrim(I) all: 1.156 / % possible all: 8.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.22→48.91 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.936 / SU B: 7.127 / SU ML: 0.171 / Cross valid method: THROUGHOUT / ESU R Free: 0.368 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.657 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.22→48.91 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|