| 登録情報 | データベース: PDB / ID: 8in6
|
|---|
| タイトル | Eisenia hydrolysis-enhancing protein from Aplysia kurodai complex with tannic acid |
|---|
 要素 要素 | 25 kDa polyphenol-binding protein |
|---|
 キーワード キーワード | UNKNOWN FUNCTION / protect glucosidase from inhibition |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
: / Chitin-binding domain type 2 / Chitin binding domain / Chitin binding Peritrophin-A domain / Chitin-binding type-2 domain profile. / Chitin binding domain superfamily類似検索 - ドメイン・相同性 BETA-1,2,3,4,6-PENTA-O-GALLOYL-D-GLUCOPYRANOSE / 25 kDa polyphenol-binding protein類似検索 - 構成要素 |
|---|
| 生物種 |   Aplysia kurodai (アメフラシ) Aplysia kurodai (アメフラシ) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.9 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.9 Å |
|---|
 データ登録者 データ登録者 | Sun, X.M. / Ye, Y.X. / Kato, K. / Yu, J. / Yao, M. |
|---|
| 資金援助 |  日本, 3件 日本, 3件 | 組織 | 認可番号 | 国 |
|---|
| Japan Society for the Promotion of Science (JSPS) | 21H01754 | 日本 | | Japan Agency for Medical Research and Development (AMED) | JP18am0101071 | 日本 | | Japan Agency for Medical Research and Development (AMED) | JP19am0101083 | 日本 |
|
|---|
 引用 引用 | ジャーナル: Elife / 年: 2023
タイトル: Structural basis of EHEP-mediated offense against phlorotannin-induced defense from brown algae to protect aku BGL activity.
著者: Sun, X. / Ye, Y. / Sakurai, N. / Wang, H. / Kato, K. / Yu, J. / Yuasa, K. / Tsuji, A. / Yao, M. |
|---|
| 履歴 | | 登録 | 2023年3月8日 | 登録サイト: PDBJ / 処理サイト: PDBJ |
|---|
| 改定 1.0 | 2023年11月15日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2024年11月13日 | Group: Structure summary
カテゴリ: pdbx_entry_details / pdbx_modification_feature
Item: _pdbx_entry_details.has_protein_modification |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード


 PDBj
PDBj 集合体
集合体
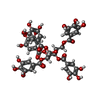
 分子量: 940.677 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C41H32O26 / タイプ: SUBJECT OF INVESTIGATION
分子量: 940.677 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C41H32O26 / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 85 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 85 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析