 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8hi6: Crystal structure of the NADP+ and MSA bound N terminal domain of... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8hi6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the NADP+ and MSA bound N terminal domain of bi-functional malonyl-CoA reductase from Roseiflexus castenholzii | ||||||
 Components Components | Short-chain dehydrogenase/reductase SDR | ||||||
 Keywords Keywords | OXIDOREDUCTASE / The 3-hydroxypropionate cycle / Short chain dehydrogenase | ||||||
| Function / homology |  Function and homology information Function and homology informationfatty acid elongation / oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor / nucleotide binding Similarity search - Function | ||||||
| Biological species |  Roseiflexus castenholzii DSM 13941 (bacteria) Roseiflexus castenholzii DSM 13941 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Zhang, X. / Wu, W.P. / Xu, X. | ||||||
| Funding support |  China, 1items China, 1items
| ||||||
 Citation Citation | Journal: mBio / Year: 2023 Title: Structural basis of a bi-functional malonyl-CoA reductase (MCR) from the photosynthetic green non-sulfur bacterium . Authors: Xin Zhang / Jiyu Xin / Zhiguo Wang / Wenping Wu / Yutong Liu / Zhenzhen Min / Yueyong Xin / Bing Liu / Jun He / Xingwei Zhang / Xiaoling Xu / Abstract: Malonyl-CoA reductase (MCR) is a NADPH-dependent bi-functional enzyme that performs alcohol dehydrogenase and aldehyde dehydrogenase (CoA-acylating) activities in the N- and C-terminal fragments, ...Malonyl-CoA reductase (MCR) is a NADPH-dependent bi-functional enzyme that performs alcohol dehydrogenase and aldehyde dehydrogenase (CoA-acylating) activities in the N- and C-terminal fragments, respectively. It catalyzes the two-step reduction of malonyl-CoA to 3-hydroxypropionate (3-HP), a key reaction in the autotrophic CO fixation cycles of green non-sulfur bacteria and the archaea . However, the structural basis underlying substrate selection, coordination, and the subsequent catalytic reactions of full-length MCR is largely unknown. For the first time, we here determined the structure of full-length MCR from the photosynthetic green non-sulfur bacterium (MCR) at 3.35 Å resolution. Furthermore, we determined the crystal structures of the N- and C-terminal fragments bound with reaction intermediates NADP and malonate semialdehyde (MSA) at 2.0 Å and 2.3 Å, respectively, and elucidated the catalytic mechanisms using a combination of molecular dynamics simulations and enzymatic analyses. Full-length MCR was a homodimer of two cross-interlocked subunits, each containing four tandemly arranged short-chain dehydrogenase/reductase (SDR) domains. Only the catalytic domains SDR1 and SDR3 incorporated additional secondary structures that changed with NADP-MSA binding. The substrate, malonyl-CoA, was immobilized in the substrate-binding pocket of SDR3 through coordination with Arg1164 and Arg799 of SDR4 and the extra domain, respectively. Malonyl-CoA was successively reduced through protonation by the Tyr743-Arg746 pair in SDR3 and the catalytic triad (Thr165-Tyr178-Lys182) in SDR1 after nucleophilic attack from NADPH hydrides. IMPORTANCE The bi-functional MCR catalyzes NADPH-dependent reduction of malonyl-CoA to 3-HP, an important metabolic intermediate and platform chemical, from biomass. The individual MCR-N and MCR-C fragments, which contain the alcohol dehydrogenase and aldehyde dehydrogenase (CoA-acylating) activities, respectively, have previously been structurally investigated and reconstructed into a malonyl-CoA pathway for the biosynthetic production of 3-HP. However, no structural information for full-length MCR has been available to illustrate the catalytic mechanism of this enzyme, which greatly limits our capacity to increase the 3-HP yield of recombinant strains. Here, we report the cryo-electron microscopy structure of full-length MCR for the first time and elucidate the mechanisms underlying substrate selection, coordination, and catalysis in the bi-functional MCR. These findings provide a structural and mechanistic basis for enzyme engineering and biosynthetic applications of the 3-HP carbon fixation pathways. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8hi6.cif.gz | 124.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8hi6.ent.gz | 92.1 KB | Display | PDB format |
| PDBx/mmJSON format | 8hi6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hi/8hi6ftp://data.pdbj.org/pub/pdb/validation_reports/hi/8hi6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8hi4SC  8hi5C C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 63446.832 Da / Num. of mol.: 1 / Fragment: N terminal domain Source method: isolated from a genetically manipulated source Details: His573-His578 are expression tag used for purification of the target protein Source: (gene. exp.) Roseiflexus castenholzii DSM 13941 (bacteria)Strain: DSM 13941 / HLO8 / Gene: Rcas_2929 / Production host: |
|---|---|
| #2: Chemical | ChemComp-FK2 / 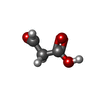  Mass: 88.062 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 88.062 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O3 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-NAP /   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 / Feature type: SUBJECT OF INVESTIGATION |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 226 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 226 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.66 Å3/Da / Density % sol: 66.37 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop Details: 1 M sodium malonate pH 5.0, 0.1 M sodium acetate trihydrate pH 4.5, and 3% PEG 20,000 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL10U2 / Wavelength: 0.97915 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Oct 30, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97915 Å / Relative weight: 1 |
| Reflection | Resolution: 2→44.91 Å / Num. obs: 65288 / % possible obs: 99.92 % / Redundancy: 38 % / CC1/2: 0.999 / Net I/σ(I): 19.95 |
| Reflection shell | Resolution: 2→2.03 Å / Num. unique obs: 2754 / CC1/2: 0.813 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 8HI4 Resolution: 2→44.91 Å / SU ML: 0.18 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 20.08 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 80.5 Å2 / Biso mean: 32.1632 Å2 / Biso min: 16.52 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→44.91 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 23
|
