 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8grt: Small Dipeptide Analogues developed by Co-crystal Structure of St... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8grt | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Small Dipeptide Analogues developed by Co-crystal Structure of Stenotrophomonas maltophilia Dipeptidyl Peptidase 7 | ||||||||||||||||||||||||
 Components Components | Dipeptidyl-peptidase | ||||||||||||||||||||||||
 Keywords Keywords | HYDROLASE / S46 / DPP / aminopeptidase / antibiotics / NFGNR / NFGNB / sepsis / pneumonia | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on peptide bonds (peptidases); Dipeptidyl-peptidases and tripeptidyl-peptidases / serine-type aminopeptidase activity / dipeptidyl-peptidase activity / peptide catabolic process / proteolysis Similarity search - Function | ||||||||||||||||||||||||
| Biological species |  Stenotrophomonas maltophilia (bacteria) Stenotrophomonas maltophilia (bacteria) | ||||||||||||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.59 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.59 Å | ||||||||||||||||||||||||
 Authors Authors | Yasumitsu, S. / Koushi, H. / Akihiro, N. / Yoshiyuki, Y. / Wataru, O. / Mizuki, S. / Saori, R. / Nobutada, T. / Anna, M. / Keiko, H. / Tsuda, Y. | ||||||||||||||||||||||||
| Funding support |  Japan, 7items Japan, 7items
| ||||||||||||||||||||||||
 Citation Citation | Journal: To Be Published Title: Small Dipeptide Analogues Generated by Co-crystal Structure of Bacterial Dipeptidyl Peptidase 7 to Defeat Stenotrophomonas maltophilia Authors: Koushi, H. / Yasumitsu, S. / Akihiro, N. / Suzuki, Y. / Wataru, O. / Mizuki, S. / Chisato, K. / Saori, R. / Nobutada, T. / Anna, M. / Keiko, H. / Yuko, Y. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8grt.cif.gz | 286.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8grt.ent.gz | 222.2 KB | Display | PDB format |
| PDBx/mmJSON format | 8grt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gr/8grtftp://data.pdbj.org/pub/pdb/validation_reports/gr/8grt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7dkbS S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 78061.461 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Stenotrophomonas maltophilia (strain R551-3) (bacteria)Strain: R551-3 / Cell: Bacteria / Gene: Smal_0807 / Plasmid: pET22b Details (production host): Full synthetic and codon optimised Production host: References: UniProt: B4SLK2, Hydrolases; Acting on peptide bonds (peptidases); Dipeptidyl-peptidases and tripeptidyl-peptidases #2: Chemical | 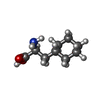  Type: L-peptide linking / Mass: 171.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO2 / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 171.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | 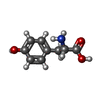  Type: L-peptide linking / Mass: 181.189 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H11NO3 Type: L-peptide linking / Mass: 181.189 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H11NO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 51.71 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8.5 / Details: 20% PEG 8000, 200mM Amm. Acetate, 20% Glycerol |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: nitrogen stream / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8 / Beamline: BL44XU / Wavelength: 0.9 Å |
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Oct 26, 2016 |
| Radiation | Monochromator: SILICON CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 2.59→34.58 Å / Num. obs: 47008 / % possible obs: 99 % / Redundancy: 3.7 % / Biso Wilson estimate: 35.788 Å2 / CC1/2: 0.994 / Rmerge(I) obs: 0.109 / Rpim(I) all: 0.066 / Rrim(I) all: 0.128 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 2.59→2.63 Å / Redundancy: 2.1 % / Rmerge(I) obs: 0.565 / Num. unique obs: 1999 / CC1/2: 0.727 / Rpim(I) all: 0.361 / % possible all: 84.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 7DKB Resolution: 2.59→30.002 Å / Cor.coef. Fo:Fc: 0.951 / Cor.coef. Fo:Fc free: 0.907 / SU B: 11.509 / SU ML: 0.24 / Cross valid method: FREE R-VALUE / ESU R: 1.486 / ESU R Free: 0.317 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.889 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.59→30.002 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
|
