 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8gkz: Human mitochondrial serine hydroxymethyltransferase (SHMT2) Y105F... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8gkz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human mitochondrial serine hydroxymethyltransferase (SHMT2) Y105F in complex with PLP, glycine and AGF362 inhibitor | ||||||
 Components Components | Serine hydroxymethyltransferase, mitochondrial | ||||||
 Keywords Keywords | TRANSFERASE/INHIBITOR / SHMT2 mutant / tetramer / glycine synthesis / inhibitor / TRANSFERASE / TRANSFERASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationformate biosynthetic process / hydroxytrimethyllysine aldolase activity / BRISC complex / glycine metabolic process / L-allo-threonine aldolase activity / regulation of mitochondrial translation / response to vitamin B6 / regulation of oxidative phosphorylation / L-serine metabolic process / L-serine biosynthetic process ...formate biosynthetic process / hydroxytrimethyllysine aldolase activity / BRISC complex / glycine metabolic process / L-allo-threonine aldolase activity / regulation of mitochondrial translation / response to vitamin B6 / regulation of oxidative phosphorylation / L-serine metabolic process / L-serine biosynthetic process / glycine hydroxymethyltransferase / glycine hydroxymethyltransferase activity / : / Metabolism of folate and pterines / tetrahydrofolate metabolic process / response to type I interferon / tetrahydrofolate interconversion / protein K63-linked deubiquitination / regulation of aerobic respiration / amino acid binding / mitochondrial nucleoid / RHOG GTPase cycle / one-carbon metabolic process / Mitochondrial protein degradation / protein tetramerization / pyridoxal phosphate binding / protein homotetramerization / mitochondrial inner membrane / mitochondrial matrix / positive regulation of cell population proliferation / chromatin binding / mitochondrion / extracellular exosome / identical protein binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.75 Å | ||||||
 Authors Authors | Katinas, J.M. / Dann III, C.E. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Biochemistry / Year: 2024 Title: Structural Characterization of 5-Substituted Pyrrolo[3,2- d ]pyrimidine Antifolate Inhibitors in Complex with Human Serine Hydroxymethyl Transferase 2. Authors: Katinas, J.M. / Nayeen, M.J. / Schneider, M. / Shah, K. / Fifer, A.N. / Klapper, L.M. / Sharma, A. / Thalluri, K. / Van Nieuwenhze, M.S. / Hou, Z. / Gangjee, A. / Matherly, L.H. / Dann 3rd, C.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8gkz.cif.gz | 361.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8gkz.ent.gz | 294.1 KB | Display | PDB format |
| PDBx/mmJSON format | 8gkz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gk/8gkzftp://data.pdbj.org/pub/pdb/validation_reports/gk/8gkz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8gksC  8gktC  8gkuC  8gkwC  8gkyC  8t4oC  8t4pC  8tlcC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 54589.926 Da / Num. of mol.: 2 / Mutation: Y105F Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SHMT2 / Production host:  References: UniProt: P34897, glycine hydroxymethyltransferase #2: Chemical | 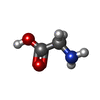  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5NO2#3: Chemical |   Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#4: Chemical | ChemComp-Y72 / | 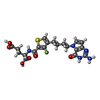  Mass: 479.482 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22FN5O6S / Feature type: SUBJECT OF INVESTIGATION Mass: 479.482 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22FN5O6S / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 20 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 20 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.43 Å3/Da / Density % sol: 64.13 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: batch mode / pH: 7.5 Details: 20 mM sodium phosphate pH 7.5, 100 mM NaCl, 0.2 mM EDTA, and 0.5 mM TCEP and PLP loaded His-SHMT2 concentrated to 0.01 to 0.02 mM |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 4.2.2 / Wavelength: 1 Å |
| Detector | Type: RDI CMOS_8M / Detector: CMOS / Date: Jun 23, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.61→48.74 Å / Num. obs: 46865 / % possible obs: 99.1 % / Redundancy: 9.7 % / CC1/2: 0.95 / Rmerge(I) obs: 0.177 / Rpim(I) all: 0.062 / Rrim(I) all: 0.189 / Net I/σ(I): 13.7 |
| Reflection shell | Resolution: 2.61→2.7 Å / Rmerge(I) obs: 3.337 / Mean I/σ(I) obs: 0.8 / Num. unique obs: 4154 / CC1/2: 0.47 / Rpim(I) all: 2.006 / Rrim(I) all: 3.967 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.75→48.736 Å / SU ML: 0.35 / Cross valid method: THROUGHOUT / σ(F): 1.33 / Phase error: 26.44 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→48.736 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -18.7681 Å / Origin y: 65.107 Å / Origin z: 6.5414 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
