 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8g50: E. coli DHFR complex with NADP+ and folate: EF-X excited state mo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8g50 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | E. coli DHFR complex with NADP+ and folate: EF-X excited state model by Laue diffraction (electric field along b axis; 8-fold extrapolation of structure factor differences) | |||||||||||||||
 Components Components | Dihydrofolate reductase | |||||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / Dihydrofolate Reductase | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationmethotrexate binding / dihydrofolic acid binding / 10-formyltetrahydrofolate biosynthetic process / response to methotrexate / folic acid biosynthetic process / folic acid binding / NADP+ binding / dihydrofolate metabolic process / dihydrofolate reductase / dihydrofolate reductase activity ...methotrexate binding / dihydrofolic acid binding / 10-formyltetrahydrofolate biosynthetic process / response to methotrexate / folic acid biosynthetic process / folic acid binding / NADP+ binding / dihydrofolate metabolic process / dihydrofolate reductase / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / NADPH binding / one-carbon metabolic process / NADP binding / response to xenobiotic stimulus / response to antibiotic / cytosol Similarity search - Function | |||||||||||||||
| Biological species |  | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.7 Å | |||||||||||||||
 Authors Authors | Greisman, J.B. / Dalton, K.M. / Brookner, D.E. / Klureza, M.A. / Hekstra, D.R. | |||||||||||||||
| Funding support |  United States, 4items United States, 4items
| |||||||||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2024 Title: Perturbative diffraction methods resolve a conformational switch that facilitates a two-step enzymatic mechanism. Authors: Greisman, J.B. / Dalton, K.M. / Brookner, D.E. / Klureza, M.A. / Sheehan, C.J. / Kim, I.S. / Henning, R.W. / Russi, S. / Hekstra, D.R. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8g50.cif.gz | 237.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8g50.ent.gz | 171.6 KB | Display | PDB format |
| PDBx/mmJSON format | 8g50.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g5/8g50ftp://data.pdbj.org/pub/pdb/validation_reports/g5/8g50 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5sssC  5sstC  5ssuC  5ssvC  5sswC  7fplC  7fpmC  7fpnC  7fpoC  7fppC  7fpqC  7fprC  7fpsC  7fptC  7fpuC  7fpvC  7fpwC  7fpxC  7fpyC  7fpzC  7fq0C  7fq1C  7fq2C  7fq3C  7fq4C  7fq5C  7fq6C  7fq7C  7fq8C  7fq9C  7fqaC  7fqbC  7fqcC  7fqdC  7fqeC  7fqfC  7fqgC  8daiC  8g4zC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18051.338 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 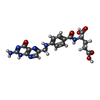  Mass: 441.397 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H19N7O6 Mass: 441.397 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H19N7O6#3: Chemical |   Mass: 743.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H28N7O17P3#4: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.53 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop Details: 20 mM imadazole (pH 5.4-5.8), 15-20% PEG 400, 125 mM MnCl2 PH range: 5.4 - 5.8 |
-Data collection
| Diffraction | Mean temperature: 278 K / Serial crystal experiment: N | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 14-ID-B / Wavelength: 1.02 - 1.18 | ||||||||||||||||||||||||
| Detector | Type: RAYONIX MX340-HS / Detector: CCD / Date: Apr 2, 2021 | ||||||||||||||||||||||||
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||
| Reflection | Entry-ID: 8G50 / CC star: 0.997 / Diffraction-ID: 1 / % possible obs: 99.4 %
| ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / % possible all: 99.5 / Resolution: 1.7→1.76 Å
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.7→41.36 Å / SU ML: 0.3158 / Cross valid method: FREE R-VALUE / σ(F): 0 / Phase error: 37.698 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 5.54 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→41.36 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 14.9732027231 Å / Origin y: 34.1065930665 Å / Origin z: 24.8020526262 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
