 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8doc: Crystal structure of RPE65 in complex with compound 16e and palmitate -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8doc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of RPE65 in complex with compound 16e and palmitate | ||||||
 Components Components | Retinoid isomerohydrolase | ||||||
 Keywords Keywords | HYDROLASE / 7-BLADED BETA PROPELLER / MONOTOPIC MEMBRANE PROTEIN / NON-HEME IRON ENZYME / RETINOID ISOMERASE / INHIBITOR / COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationretinoid isomerohydrolase / lutein isomerase / retinol isomerase activity / all-trans-retinyl-palmitate hydrolase, 11-cis retinol forming activity / all-trans-retinyl-ester hydrolase, 11-cis retinol forming activity / zeaxanthin biosynthetic process / beta-carotene 15,15'-dioxygenase activity / The canonical retinoid cycle in rods (twilight vision) / detection of light stimulus involved in visual perception / retinal metabolic process ...retinoid isomerohydrolase / lutein isomerase / retinol isomerase activity / all-trans-retinyl-palmitate hydrolase, 11-cis retinol forming activity / all-trans-retinyl-ester hydrolase, 11-cis retinol forming activity / zeaxanthin biosynthetic process / beta-carotene 15,15'-dioxygenase activity / The canonical retinoid cycle in rods (twilight vision) / detection of light stimulus involved in visual perception / retinal metabolic process / phosphatidylcholine binding / cardiolipin binding / retina homeostasis / phosphatidylserine binding / endoplasmic reticulum membrane / metal ion binding / identical protein binding / membrane / nucleus / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.1 Å | ||||||
 Authors Authors | Bassetto, M. / Kiser, P.D. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2023 Title: Tuning the Metabolic Stability of Visual Cycle Modulators through Modification of an RPE65 Recognition Motif. Authors: Bassetto, M. / Zaluski, J. / Li, B. / Zhang, J. / Badiee, M. / Kiser, P.D. / Tochtrop, G.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8doc.cif.gz | 128.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8doc.ent.gz | 95.8 KB | Display | PDB format |
| PDBx/mmJSON format | 8doc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/do/8docftp://data.pdbj.org/pub/pdb/validation_reports/do/8doc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ryxS S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 60935.039 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) References: UniProt: Q28175, retinoid isomerohydrolase, lutein isomerase |
|---|
-Non-polymers , 5 types, 350 molecules 


| #2: Chemical | ChemComp-FE2 /  Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION |
|---|---|
| #3: Chemical | ChemComp-CXS /  Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM |
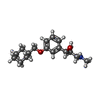
| #4: Chemical | ChemComp-PLM /  Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION |
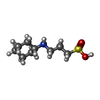
| #5: Chemical | ChemComp-SYL / ( Mass: 277.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H27NO2 / Feature type: SUBJECT OF INVESTIGATION Mass: 277.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H27NO2 / Feature type: SUBJECT OF INVESTIGATION |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 346 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.19 Å3/Da / Density % sol: 61.46 % |
|---|---|
| Crystal grow | Temperature: 281 K / Method: vapor diffusion, hanging drop / pH: 10.5 Details: 30% PEG 400, 100 mM CAPS, pH 10.5, 500 mM ammonium sulfate, 10% v/v |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-E / Wavelength: 0.9791 Å / Beamline: 24-ID-E / Wavelength: 0.9791 Å |
| Detector | Type: ADSC HF-4M / Detector: PIXEL / Date: Mar 7, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9791 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→50 Å / Num. obs: 46592 / % possible obs: 99.9 % / Redundancy: 12.7 % / CC1/2: 0.998 / Rmerge(I) obs: 0.176 / Net I/σ(I): 10.33 |
| Reflection shell | Resolution: 2.1→2.23 Å / Rmerge(I) obs: 2.338 / Mean I/σ(I) obs: 1.09 / Num. unique obs: 7529 / CC1/2: 0.399 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 4ryx Resolution: 2.1→48.05 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.957 / SU B: 5.415 / SU ML: 0.129 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.156 / ESU R Free: 0.143 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 1 Å / Shrinkage radii: 1 Å / VDW probe radii: 1.3 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 108.6 Å2 / Biso mean: 41.965 Å2 / Biso min: 27.65 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.1→48.05 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.154 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
