| 登録情報 | データベース: PDB / ID: 8cjj
|
|---|
| タイトル | Crystal structure of human tryptophan hydroxylase 1 in complex with inhibitor KM-06-057 |
|---|
 要素 要素 | Tryptophan 5-hydroxylase 1 |
|---|
 キーワード キーワード | METAL BINDING PROTEIN / catalytic domain of human tryptophan hydroxylase 1 (TPH1) / inhibitor complex |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
regulation of hemostasis / : / tryptophan 5-monooxygenase / tryptophan 5-monooxygenase activity / Serotonin and melatonin biosynthesis / serotonin biosynthetic process / platelet degranulation / NGF-stimulated transcription / bone remodeling / mammary gland alveolus development ...regulation of hemostasis / : / tryptophan 5-monooxygenase / tryptophan 5-monooxygenase activity / Serotonin and melatonin biosynthesis / serotonin biosynthetic process / platelet degranulation / NGF-stimulated transcription / bone remodeling / mammary gland alveolus development / positive regulation of fat cell differentiation / neuron projection / iron ion binding / cytosol類似検索 - 分子機能 Tryptophan 5-monooxygenase / Tryptophan 5-hydroxylase, catalytic domain / Tyrosine 3-monooxygenase-like / Aromatic amino acid hydroxylase, iron/copper binding site / Biopterin-dependent aromatic amino acid hydroxylases signature. / Aromatic amino acid hydroxylase / Aromatic amino acid hydroxylase, C-terminal / Aromatic amino acid monoxygenase, C-terminal domain superfamily / Aromatic amino acid hydroxylase superfamily / Biopterin-dependent aromatic amino acid hydroxylase ...Tryptophan 5-monooxygenase / Tryptophan 5-hydroxylase, catalytic domain / Tyrosine 3-monooxygenase-like / Aromatic amino acid hydroxylase, iron/copper binding site / Biopterin-dependent aromatic amino acid hydroxylases signature. / Aromatic amino acid hydroxylase / Aromatic amino acid hydroxylase, C-terminal / Aromatic amino acid monoxygenase, C-terminal domain superfamily / Aromatic amino acid hydroxylase superfamily / Biopterin-dependent aromatic amino acid hydroxylase / Biopterin-dependent aromatic amino acid hydroxylase family profile. / ACT domain profile. / ACT domain / ACT-like domain類似検索 - ドメイン・相同性 |
|---|
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.66415657298 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.66415657298 Å |
|---|
 データ登録者 データ登録者 | Schuetz, A. / Mallow, K. / Nazare, M. / Specker, E. / Heinemann, U. |
|---|
| 資金援助 |  ドイツ, 1件 ドイツ, 1件 | 組織 | 認可番号 | 国 |
|---|
| Helmholtz Association | | ドイツ |
|
|---|
 引用 引用 | ジャーナル: J.Med.Chem. / 年: 2023
タイトル: Structure-Based Design of Xanthine-Imidazopyridines and -Imidazothiazoles as Highly Potent and In Vivo Efficacious Tryptophan Hydroxylase Inhibitors.
著者: Specker, E. / Wesolowski, R. / Schutz, A. / Matthes, S. / Mallow, K. / Wasinska-Kalwa, M. / Winkler, L. / Oder, A. / Alenina, N. / Pleimes, D. / von Kries, J.P. / Heinemann, U. / Bader, M. / Nazare, M. |
|---|
| 履歴 | | 登録 | 2023年2月13日 | 登録サイト: PDBE / 処理サイト: PDBE |
|---|
| 改定 1.0 | 2024年1月10日 | Provider: repository / タイプ: Initial release |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード





 PDBj
PDBj



 集合体
集合体


 分子量: 55.845 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Fe
分子量: 55.845 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Fe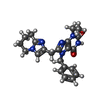
 分子量: 404.465 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C22H24N6O2 / タイプ: SUBJECT OF INVESTIGATION
分子量: 404.465 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C22H24N6O2 / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 265 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 265 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析