| Entry | Database: PDB / ID: 8byj
|
|---|
| Title | The structures of Ace2 in complex with bicyclic peptide inhibitor |
|---|
 Components Components | - ALA-CYS-VAL-ARG-SER-HIS-CYS-SER-SER-LEU-LEU-PRO-ARG-ILE-HIS-CYS-ALA
- Processed angiotensin-converting enzyme 2
|
|---|
 Keywords Keywords | MEMBRANE PROTEIN / Cov-2-sars bind proteins |
|---|
| Function / homology |  Function and homology information Function and homology information
positive regulation of amino acid transport / angiotensin-converting enzyme 2 / positive regulation of L-proline import across plasma membrane / Hydrolases; Acting on peptide bonds (peptidases); Metallocarboxypeptidases / angiotensin-mediated drinking behavior / positive regulation of gap junction assembly / tryptophan transport / maternal process involved in female pregnancy / regulation of systemic arterial blood pressure by renin-angiotensin / regulation of cardiac conduction ...positive regulation of amino acid transport / angiotensin-converting enzyme 2 / positive regulation of L-proline import across plasma membrane / Hydrolases; Acting on peptide bonds (peptidases); Metallocarboxypeptidases / angiotensin-mediated drinking behavior / positive regulation of gap junction assembly / tryptophan transport / maternal process involved in female pregnancy / regulation of systemic arterial blood pressure by renin-angiotensin / regulation of cardiac conduction / transporter activator activity / amino acid import across plasma membrane / peptidyl-dipeptidase activity / regulation of vasoconstriction / Metabolism of Angiotensinogen to Angiotensins / carboxypeptidase activity / angiotensin maturation / receptor-mediated endocytosis of virus by host cell / viral life cycle / Attachment and Entry / metallocarboxypeptidase activity / positive regulation of cardiac muscle contraction / regulation of cytokine production / blood vessel diameter maintenance / negative regulation of smooth muscle cell proliferation / brush border membrane / negative regulation of ERK1 and ERK2 cascade / positive regulation of reactive oxygen species metabolic process / metallopeptidase activity / endocytic vesicle membrane / regulation of cell population proliferation / virus receptor activity / regulation of inflammatory response / cilium / endopeptidase activity / Induction of Cell-Cell Fusion / Potential therapeutics for SARS / membrane fusion / Attachment and Entry / entry receptor-mediated virion attachment to host cell / receptor-mediated virion attachment to host cell / apical plasma membrane / membrane raft / endoplasmic reticulum lumen / symbiont entry into host cell / negative regulation of transcription by RNA polymerase II / cell surface / : / extracellular exosome / extracellular region / zinc ion binding / membrane / identical protein binding / plasma membraneSimilarity search - Function Collectrin domain / Renal amino acid transporter / Collectrin-like domain profile. / Peptidase M2, peptidyl-dipeptidase A / Angiotensin-converting enzyme / Peptidase family M2 domain profile. / Neutral zinc metallopeptidases, zinc-binding region signature.Similarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human)
synthetic construct (others) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.07 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.07 Å |
|---|
 Authors Authors | Brear, P. / Lulla, A. / Harman, M. / Dods, R. / Chen, L. / Bezerra, G. / Demydchuk, Y. / Stanway, S. / Hyvonen, M. |
|---|
| Funding support | 1items | Organization | Grant number | Country |
|---|
| Other private | | |
|
|---|
 Citation Citation | Journal: J.Med.Chem. / Year: 2023
Title: Structure-Guided Chemical Optimization of Bicyclic Peptide ( Bicycle ) Inhibitors of Angiotensin-Converting Enzyme 2.
Authors: Harman, M.A.J. / Stanway, S.J. / Scott, H. / Demydchuk, Y. / Bezerra, G.A. / Pellegrino, S. / Chen, L. / Brear, P. / Lulla, A. / Hyvonen, M. / Beswick, P.J. / Skynner, M.J. |
|---|
| History | | Deposition | Dec 13, 2022 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Sep 20, 2023 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 20, 2024 | Group: Structure summary / Category: pdbx_entry_details / pdbx_modification_feature / Item: _pdbx_entry_details.has_protein_modification |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj


 Assembly
Assembly





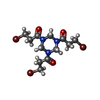

 Mass: 65.409 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: Zn Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 492.002 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18Br3N3O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 492.002 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18Br3N3O3 / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation / Beamline: I04 / Wavelength: 0.9795 Å
/ Beamline: I04 / Wavelength: 0.9795 Å Processing
Processing