 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8axz: Crystal structure of human methionine adenosyltransferase 2A (MAT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8axz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human methionine adenosyltransferase 2A (MAT2A) in complex with S-adenosylmethionine, adenosin and diphosphono-aminophosphonic acid. | ||||||
 Components Components | S-adenosylmethionine synthase isoform type-2 | ||||||
 Keywords Keywords | TRANSFERASE / methionine adenosyltransferase / SAM-producing metabolic enzyme / cancer target | ||||||
| Function / homology |  Function and homology information Function and homology informationmethionine adenosyltransferase complex / methionine adenosyltransferase / methionine adenosyltransferase activity / protein heterooligomerization / S-adenosylmethionine biosynthetic process / Methylation / cellular response to methionine / protein hexamerization / small molecule binding / one-carbon metabolic process ...methionine adenosyltransferase complex / methionine adenosyltransferase / methionine adenosyltransferase activity / protein heterooligomerization / S-adenosylmethionine biosynthetic process / Methylation / cellular response to methionine / protein hexamerization / small molecule binding / one-carbon metabolic process / positive regulation of TORC1 signaling / ATP binding / metal ion binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.154 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.154 Å | ||||||
 Authors Authors | Nawrotek, A. / Vuillard, L. / Miallau, L. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of human methionine adenosyltransferase 2A (MAT2A) in complex with S-adenosylmethionine, adenosin and diphosphono-aminophosphonic acid. Authors: Nawrotek, A. / Vuillard, L. / Miallau, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8axz.cif.gz | 103.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8axz.ent.gz | 75.6 KB | Display | PDB format |
| PDBx/mmJSON format | 8axz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ax/8axzftp://data.pdbj.org/pub/pdb/validation_reports/ax/8axz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2p02S S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 43921.805 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MAT2A, AMS2, MATA2 / Production host:  |
|---|
-Non-polymers , 8 types, 288 molecules 


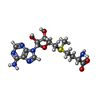



| #2: Chemical | ChemComp-PPK / ( Mass: 256.970 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H6NO9P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 256.970 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H6NO9P3 / Feature type: SUBJECT OF INVESTIGATION | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |  Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K / Feature type: SUBJECT OF INVESTIGATION Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | ChemComp-SAM / |  Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S / Feature type: SUBJECT OF INVESTIGATION Mass: 398.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H22N6O5S / Feature type: SUBJECT OF INVESTIGATION#6: Chemical | ChemComp-ADN / |  Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 / Feature type: SUBJECT OF INVESTIGATION#7: Chemical | ChemComp-PEG / |  Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 / Feature type: SUBJECT OF INVESTIGATION#8: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 279 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.2 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.12 M Alcohols (0.2 M 1,6-Hexanediol; 0.2 M 1-Butanol; 0.2 M 1,2-Propanediol; 0.2 M 2-Propanol; 0.2 M 1,4-Butanediol; 0.2 M 1,3-Propanediol), 0.1 M Buffer System 3 pH 8.5 (Tris (base); ...Details: 0.12 M Alcohols (0.2 M 1,6-Hexanediol; 0.2 M 1-Butanol; 0.2 M 1,2-Propanediol; 0.2 M 2-Propanol; 0.2 M 1,4-Butanediol; 0.2 M 1,3-Propanediol), 0.1 M Buffer System 3 pH 8.5 (Tris (base); BICINE), 50 % Precipitant mix 4 (25 % v/v MPD; 25 % PEG 1000; 25 % w/v PEG 3350) |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 0.92 Å / Beamline: I04-1 / Wavelength: 0.92 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Apr 30, 2014 / Details: MIRRORS |
| Radiation | Monochromator: SI111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 1.1→73.37 Å / Num. obs: 176512 / % possible obs: 99.1 % / Redundancy: 12.3 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 24.5 |
| Reflection shell | Resolution: 1.1→1.14 Å / Redundancy: 9.7 % / Rmerge(I) obs: 0.09 / Mean I/σ(I) obs: 3.4 / Num. unique obs: 1403 / % possible all: 99 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2P02 Resolution: 1.154→58.63 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.96 / SU R Cruickshank DPI: 0.047 / Cross valid method: THROUGHOUT / SU R Blow DPI: 0.048 / SU Rfree Blow DPI: 0.049 / SU Rfree Cruickshank DPI: 0.048
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.68 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.14 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.154→58.63 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.154→1.21 Å
|
