- PDB-7xyq: Crystal strucutre of PD-L1 and the computationally designed DBL1_... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 7xyq
Title
Crystal strucutre of PD-L1 and the computationally designed DBL1_03 protein binder
Components
CD274 molecule
DBL1_03
Keywords
IMMUNE SYSTEM / PD-L1
Function / homology
Function and homology information
positive regulation of tolerance induction to tumor cell / negative regulation of tumor necrosis factor superfamily cytokine production / positive regulation of activated CD8-positive, alpha-beta T cell apoptotic process / negative regulation of CD8-positive, alpha-beta T cell activation / negative regulation of CD4-positive, alpha-beta T cell proliferation / TRIF-dependent toll-like receptor signaling pathway / negative regulation of interleukin-10 production / negative regulation of activated T cell proliferation / negative regulation of type II interferon production / positive regulation of interleukin-10 production ...positive regulation of tolerance induction to tumor cell / negative regulation of tumor necrosis factor superfamily cytokine production / positive regulation of activated CD8-positive, alpha-beta T cell apoptotic process / negative regulation of CD8-positive, alpha-beta T cell activation / negative regulation of CD4-positive, alpha-beta T cell proliferation / TRIF-dependent toll-like receptor signaling pathway / negative regulation of interleukin-10 production / negative regulation of activated T cell proliferation / negative regulation of type II interferon production / positive regulation of interleukin-10 production / T cell costimulation / response to cytokine / positive regulation of T cell proliferation / recycling endosome membrane / actin cytoskeleton / cellular response to lipopolysaccharide / early endosome membrane / transcription coactivator activity / cell surface receptor signaling pathway / immune response / external side of plasma membrane / extracellular exosome / nucleoplasm Similarity search - Function
National Natural Science Foundation of China (NSFC)
92169208
China
Citation
Journal: Nature / Year: 2023 Title: De novo design of protein interactions with learned surface fingerprints. Authors: Pablo Gainza / Sarah Wehrle / Alexandra Van Hall-Beauvais / Anthony Marchand / Andreas Scheck / Zander Harteveld / Stephen Buckley / Dongchun Ni / Shuguang Tan / Freyr Sverrisson / Casper ...Authors: Pablo Gainza / Sarah Wehrle / Alexandra Van Hall-Beauvais / Anthony Marchand / Andreas Scheck / Zander Harteveld / Stephen Buckley / Dongchun Ni / Shuguang Tan / Freyr Sverrisson / Casper Goverde / Priscilla Turelli / Charlène Raclot / Alexandra Teslenko / Martin Pacesa / Stéphane Rosset / Sandrine Georgeon / Jane Marsden / Aaron Petruzzella / Kefang Liu / Zepeng Xu / Yan Chai / Pu Han / George F Gao / Elisa Oricchio / Beat Fierz / Didier Trono / Henning Stahlberg / Michael Bronstein / Bruno E Correia / Abstract: Physical interactions between proteins are essential for most biological processes governing life. However, the molecular determinants of such interactions have been challenging to understand, even ...Physical interactions between proteins are essential for most biological processes governing life. However, the molecular determinants of such interactions have been challenging to understand, even as genomic, proteomic and structural data increase. This knowledge gap has been a major obstacle for the comprehensive understanding of cellular protein-protein interaction networks and for the de novo design of protein binders that are crucial for synthetic biology and translational applications. Here we use a geometric deep-learning framework operating on protein surfaces that generates fingerprints to describe geometric and chemical features that are critical to drive protein-protein interactions. We hypothesized that these fingerprints capture the key aspects of molecular recognition that represent a new paradigm in the computational design of novel protein interactions. As a proof of principle, we computationally designed several de novo protein binders to engage four protein targets: SARS-CoV-2 spike, PD-1, PD-L1 and CTLA-4. Several designs were experimentally optimized, whereas others were generated purely in silico, reaching nanomolar affinity with structural and mutational characterization showing highly accurate predictions. Overall, our surface-centric approach captures the physical and chemical determinants of molecular recognition, enabling an approach for the de novo design of protein interactions and, more broadly, of artificial proteins with function.
Mass: 23757.910 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CD274 / Production host: Escherichia coli (E. coli) / References: UniProt: A0A2I2ZMM7
#2: Protein
DBL1_03
Mass: 14001.120 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: Escherichia coli (E. coli)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors China, 1items
China, 1items  Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






 PDBj
PDBj





 Assembly
Assembly

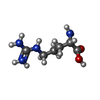
 Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N4O2
Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N4O2 Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing