登録情報 データベース : PDB / ID : 7u3hタイトル GID4 in complex with compound 7 Glucose-induced degradation protein 4 homolog キーワード / / / / / 機能・相同性 / / / / / / / / 生物種 Homo sapiens (ヒト)手法 / / / 解像度 : 1.798 Å データ登録者 Chana, C.K. / Sicheri, F. 資金援助 組織 認可番号 国 Canadian Institutes of Health Research (CIHR) FDN-143277
ジャーナル : J.Med.Chem. / 年 : 2022タイトル : Discovery and Structural Characterization of Small Molecule Binders of the Human CTLH E3 Ligase Subunit GID4.著者: Chana, C.K. / Maisonneuve, P. / Posternak, G. / Grinberg, N.G.A. / Poirson, J. / Ona, S.M. / Ceccarelli, D.F. / Mader, P. / St-Cyr, D.J. / Pau, V. / Kurinov, I. / Tang, X. / Deng, D. / Cui, W. ... 著者 : Chana, C.K. / Maisonneuve, P. / Posternak, G. / Grinberg, N.G.A. / Poirson, J. / Ona, S.M. / Ceccarelli, D.F. / Mader, P. / St-Cyr, D.J. / Pau, V. / Kurinov, I. / Tang, X. / Deng, D. / Cui, W. / Su, W. / Kuai, L. / Soll, R. / Tyers, M. / Rost, H.L. / Batey, R.A. / Taipale, M. / Gingras, A.C. / Sicheri, F. 履歴 登録 2022年2月27日 登録サイト / 処理サイト 改定 1.0 2022年10月5日 Provider / タイプ 改定 1.1 2022年10月26日 Group / カテゴリ / citation_authorItem _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation_author.identifier_ORCID 改定 1.2 2023年10月18日 Group / Refinement descriptionカテゴリ / chem_comp_bond / pdbx_initial_refinement_model
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 カナダ, 1件
カナダ, 1件  引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード







 PDBj
PDBj 集合体
集合体

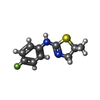
 分子量: 210.271 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H11FN2S / タイプ: SUBJECT OF INVESTIGATION
分子量: 210.271 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H11FN2S / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 86 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 86 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 24-ID-C / 波長: 0.97918 Å
/ ビームライン: 24-ID-C / 波長: 0.97918 Å 解析
解析