 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7t1p: Solution structure of 7SK stem-loop 1 with HIV-1 Tat Finland Argi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7t1p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Solution structure of 7SK stem-loop 1 with HIV-1 Tat Finland Arginine Rich Motif | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSCRIPTION / Tat / 7SK / HIV-1 | |||||||||
| Function / homology | RNA / RNA (> 10) Function and homology information Function and homology information | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) HIV-1 06TG.HT008 (virus) HIV-1 06TG.HT008 (virus) | |||||||||
| Method | SOLUTION NMR /  SOLUTION SCATTERING / simulated annealing SOLUTION SCATTERING / simulated annealing | |||||||||
 Authors Authors | Pham, V.V. / Gao, M. / D'Souza, V.M. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Commun Biol / Year: 2022 Title: A structure-based mechanism for displacement of the HEXIM adapter from 7SK small nuclear RNA. Authors: Pham, V.V. / Gao, M. / Meagher, J.L. / Smith, J.L. / D'Souza, V.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7t1p.cif.gz | 409.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7t1p.ent.gz | 342.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7t1p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t1/7t1pftp://data.pdbj.org/pub/pdb/validation_reports/t1/7t1p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7t1nC  7t1oC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|
-Links
 PDBj
PDBj






























- Assembly
Assembly
| Deposited unit | 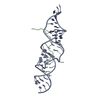
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: RNA chain | Mass: 17986.635 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) |
|---|---|
| #2: Protein/peptide | Mass: 2144.537 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HIV-1 06TG.HT008 (virus) |
-Experimental details
-Experiment
| Experiment |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR experiment |
|
- Sample preparation
Sample preparation
| Details |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample |
| ||||||||||||||||||||
| Sample conditions |
|
-Data collection
| NMR spectrometer | Type: Bruker DMX / Manufacturer: Bruker / Model: DMX / Field strength: 700 MHz |
|---|---|
| Soln scatter | Type: neutron / Buffer name: 10 mM phosphate, 70 mM NaCl, 0.1 mM EDTA / Data analysis software list: Scatter / Data reduction software list: ATSAS / Detector specific: Dectris / Detector type: Pilatus3 X 2M detector / Mean guiner radius: 2.5 nm / Num. of time frames: 10 / Sample pH: 5.6 / Source beamline: 12.3.1 / Source beamline instrument: SIBYLS / Source class: Y / Source type: 12.3.1 (SIBYLS) beam line / Temperature: 283 K |
- Processing
Processing
| NMR software |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: simulated annealing / Software ordinal: 8 | |||||||||||||||||||||||||||
| NMR representative | Selection criteria: lowest energy | |||||||||||||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 100 / Conformers submitted total number: 10 | |||||||||||||||||||||||||||
| Soln scatter model | Method: This structure was calculated with XPLOR-NIH with a CYANA input and minimized with SAXS. |
