 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7pji: Crystal structure of Pseudomonas aeruginosa guaB (IMP dehydrogena... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7pji | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Pseudomonas aeruginosa guaB (IMP dehydrogenase) bound to ATP and GDP at 1.65A resolution | ||||||
 Components Components | Inosine-5'-monophosphate dehydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / IMP dehydrogenase / guaB | ||||||
| Function / homology |  Function and homology information Function and homology informationIMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Fernandez-Justel, D. / Buey, R.M. | ||||||
| Funding support |  Spain, 1items Spain, 1items
| ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2022 Title: Diversity of mechanisms to control bacterial GTP homeostasis by the mutually exclusive binding of adenine and guanine nucleotides to IMP dehydrogenase. Authors: Fernandez-Justel, D. / Marcos-Alcalde, I. / Abascal, F. / Vidana, N. / Gomez-Puertas, P. / Jimenez, A. / Revuelta, J.L. / Buey, R.M. #1: Journal: Plant Physiol. / Year: 2021Title: Unexpected diversity of ferredoxin-dependent thioredoxin reductases in cyanobacteria. Authors: Buey, R.M. / Fernandez-Justel, D. / Gonzalez-Holgado, G. / Martinez-Julvez, M. / Gonzalez-Lopez, A. / Velazquez-Campoy, A. / Medina, M. / Buchanan, B.B. / Balsera, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7pji.cif.gz | 555.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7pji.ent.gz | 403.6 KB | Display | PDB format |
| PDBx/mmJSON format | 7pji.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pj/7pjiftp://data.pdbj.org/pub/pdb/validation_reports/pj/7pji | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7pmzC  4avfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 52049.633 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria)Gene: guaB, CAZ10_17565, CGU42_10690, CSB93_2499, DT376_00630, DY940_09800, DZ962_03005, E4V10_03770, ECC04_008545, F7O94_09425, H2O03_23015, HW05_11140, IPC1164_08420, IPC116_03070, IPC1295_13460, ...Gene: guaB, CAZ10_17565, CGU42_10690, CSB93_2499, DT376_00630, DY940_09800, DZ962_03005, E4V10_03770, ECC04_008545, F7O94_09425, H2O03_23015, HW05_11140, IPC1164_08420, IPC116_03070, IPC1295_13460, IPC1298_02025, IPC1323_03035, IPC1481_30840, IPC1505_09885, IPC1509_02385, IPC151_02390, IPC36_21115, IPC582_15390, IPC620_11170, IPC737_18950, IPC90_16110, NCTC12924_06429, NCTC13621_00582, NCTC13628_01024, PA52Ts2_5150, PAMH19_1311, RW109_RW109_02018 Production host: |
|---|
-Non-polymers , 7 types, 660 molecules 

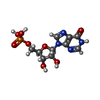




| #2: Chemical |  Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#3: Chemical |  Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM#4: Chemical |  Mass: 348.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N4O8P Mass: 348.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N4O8P#5: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#7: Chemical | ChemComp-ACT /  Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 646 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.81 Å3/Da / Density % sol: 56.26 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop Details: Condition D11 of the commercial screening Morpheus (Gorrec, 2009): 0.02M Sodium formate; 0.02M Ammonium acetate; 0.02M Sodium citrate tribasic dihydrate; 0.02M Potassium sodium tartrate ...Details: Condition D11 of the commercial screening Morpheus (Gorrec, 2009): 0.02M Sodium formate; 0.02M Ammonium acetate; 0.02M Sodium citrate tribasic dihydrate; 0.02M Potassium sodium tartrate tetrahydrate; 0.02M Sodium oxamate, 12.5% v/v MPD; 12.5% PEG 1000; 12.5% w/v PEG 3350 in 0.1M of the buffer system Tris (base), bicine, pH 8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 1.000008 Å / Beamline: I04 / Wavelength: 1.000008 Å |
| Detector | Type: DECTRIS EIGER2 XE 16M / Detector: PIXEL / Date: Feb 3, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.000008 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→50.7 Å / Num. obs: 84861 / % possible obs: 67.8 % / Redundancy: 13.9 % / Biso Wilson estimate: 25.18 Å2 / CC1/2: 0.999 / CC star: 1 / Rmerge(I) obs: 0.09 / Rpim(I) all: 0.025 / Rrim(I) all: 0.094 / Net I/σ(I): 13.18 |
| Reflection shell | Resolution: 1.65→1.71 Å / Redundancy: 13.8 % / Rmerge(I) obs: 1.76 / Mean I/σ(I) obs: 0.39 / Num. unique obs: 4228 / CC1/2: 0.287 / CC star: 0.668 / Rpim(I) all: 0.489 / Rrim(I) all: 1.827 / % possible all: 7.09 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4AVF Resolution: 1.65→50.7 Å / SU ML: 0.1407 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 26.44 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.28 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→50.7 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
