Journal: Nat Commun / Year: 2022 Title: Cryo-EM structures of a LptDE transporter in complex with Pro-macrobodies offer insight into lipopolysaccharide translocation. Authors: Mathieu Botte / Dongchun Ni / Stephan Schenck / Iwan Zimmermann / Mohamed Chami / Nicolas Bocquet / Pascal Egloff / Denis Bucher / Matilde Trabuco / Robert K Y Cheng / Janine D Brunner / ...Authors: Mathieu Botte / Dongchun Ni / Stephan Schenck / Iwan Zimmermann / Mohamed Chami / Nicolas Bocquet / Pascal Egloff / Denis Bucher / Matilde Trabuco / Robert K Y Cheng / Janine D Brunner / Markus A Seeger / Henning Stahlberg / Michael Hennig / Abstract: Lipopolysaccharides are major constituents of the extracellular leaflet in the bacterial outer membrane and form an effective physical barrier for environmental threats and for antibiotics in Gram- ...Lipopolysaccharides are major constituents of the extracellular leaflet in the bacterial outer membrane and form an effective physical barrier for environmental threats and for antibiotics in Gram-negative bacteria. The last step of LPS insertion via the Lpt pathway is mediated by the LptD/E protein complex. Detailed insights into the architecture of LptDE transporter complexes have been derived from X-ray crystallography. However, no structure of a laterally open LptD transporter, a transient state that occurs during LPS release, is available to date. Here, we report a cryo-EM structure of a partially opened LptDE transporter in complex with rigid chaperones derived from nanobodies, at 3.4 Å resolution. In addition, a subset of particles allows to model a structure of a laterally fully opened LptDE complex. Our work offers insights into the mechanism of LPS insertion, provides a structural framework for the development of antibiotics targeting LptD and describes a highly rigid chaperone scaffold to enable structural biology of challenging protein targets.
Mass: 56714.457 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: Escherichia coli (E. coli)
#2: Polysaccharide
alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose
Type: oligosaccharide, Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-maltose
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Switzerland, 1items
Switzerland, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



 PDBj
PDBj







 Assembly
Assembly


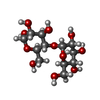

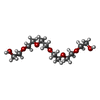
 Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM
Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM
 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 153 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 153 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing